|
|
中国第一批罕见病目录:111

有这样一群罕见病患者,因为走路不稳,摇摇摆摆,自称“企鹅”。顾大夫行医 20 余年,诊治了来自 2000 多个“企鹅”家族的患者,也见证了疾病给这些病友和家庭带来的痛苦,以及他们与疾病的不懈抗争。比如,下面的这个故事。
当幸福之家遇上SCA
文 | 君来
一个幸福的家庭该是什么模样?父亲勤劳 ,母亲贤惠,孩子聪颖可爱——这是我 13 岁前的记忆,或许也是千千万万幸福家庭的缩影。
谁知,2003 年的一次意外,打破了原本属于我们平淡而温馨的生活。我至今依然清楚地记得,那是母亲 33 岁生日前夕。那天风很大,刮落了五楼的窗玻璃,恰好砸中行经楼下的母亲。当时,鲜血流了一地……
当母亲的意识重新恢复的时候,耳朵已被缠上厚厚的纱布。大夫说,真是万幸,还差一毫米就会伤到动脉。随后,母亲卧床静养了几个月,才渐渐康复。但自此之后,母亲的平衡能力好像变差了——走路跌跌撞撞;与父亲外出散步时 ,总是控制不了地撞到他。
于是,父亲陪着母亲去附近的社区医院就医。大夫认为,母亲可能还未完全康复,建议继续休养,开了一些补充营养的维生素和安神的中成药。可母亲服药后情况没有好转,平衡能力日渐衰退,总把自己摔伤。
我们又去了附近几家综合性医院。大夫都说,是外伤导致的神经受损,可能影响到了耳蜗内部负责平衡功能的部位。母亲被收入院,接受针对神经损伤的治疗。然而,奇怪的症状没有丝毫缓解,平衡能力逐步丧失,母亲还时常感到十分头晕。
2003 年底,我们辗转来到北京的一家知名三甲医院。一位神经内科的老专家为母亲做了一系列量表检查,接着又详细询问起家族史来。母亲回忆起,我的姥爷也曾有过类似的症状,但老人家当时住在农村,过世也早,并不清楚是得了什么病。
完成所有的检查后,老专家说了一句令我们至今难以忘怀的话:“这是小脑萎缩引起了共济失调,是遗传病,治不好,不用看了。”
父亲当时就很困惑——现代医学这么发达,怎么会有看不好的病?于是他自学上网,在互联网上搜索关于小脑萎缩症的各种信息,无意间找到了顾卫红大夫的信息。
2004 年,我们在北京中日友好医院第一次见到顾大夫。经过基因检测,顾大夫诊断母亲的病确实是脊髓小脑性共济失调(spinocerebellar ataxia,SCA)。这是一类会逐渐丧失运动能力的神经系统遗传病,共有几十种亚型,母亲所患的正是最常见的 SCA3 型。
历时一年多的辗转求医,母亲的怪病终于得到了确诊,但我们一点儿也高兴不起来。因为脊髓小脑性共济失调还没有治愈的方法,只能依靠药物改善一些症状,例如头晕、头痛、失眠、肌张力过高等。患者需要长期坚持康复训练来延缓病情的进展。
确诊后,我们召开家庭会议,达成了共识:母亲从此不再外出工作,安心在家进行康复训练。为了保护日间行动不便的母亲,我们对家里进行了安全改造,装上无障碍设施。
在全家人的悉心照料下,起初母亲的病情进展比较缓慢——尽管不平稳,但可以自己行走,还可以练习站桩、太极拳、五禽戏等传统保健功法,可是病情还是在不断发展。2014 年,母亲需要扶着助行器才能行走;2017 年,肌肉进一步萎缩,腿部再也无法支撑身体,不得不使用轮椅;最近一两年,母亲已完全卧床,需要专人照料。
我眼睁睁地看着病痛日益侵蚀着母亲的身体,从健康一步一步陷入瘫痪,我倍感煎熬却无能为力。
更让我痛苦的是,SCA 像一个诅咒笼罩着我们整个家族——舅舅也出现了相同的症状,而我也被证实携带着 SCA 的致病基因,这意味着终有一天我会重复母亲的命运。但不知道身体里的这枚不定时炸弹将何时引爆,使我陷入了深深的抑郁之中。
幸有顾大夫是国内共济失调方面的专家,十余年如一日地奋斗在 SCA 诊疗的第一线,也真切关心着患者们的生活。自从确诊以来,顾大夫一直追踪母亲的病情发展,并指导治疗。
我们从最初的医患关系逐渐成为亦师亦友的好朋友。经过与顾大夫的交流和长时间的自我疏导,我渐渐走出了阴霾,开始积极面对人生。上大学的时候,我放弃了原本喜欢的计算机专业,转而学习中医,希望能让家人、今后的自己以及更多的病友少些痛苦。
其实,全国还有数以万计的家庭也在遭受着 SCA 带来的痛苦。患者发病时大多在 20 - 40 岁之间,正是建设家庭打拼事业的黄金时期。有时,一家几代人都被病魔缠身。许多病友生活在农村,没有良好的医疗条件,被长期误诊。
近年来,中国社会各方开始关注罕见病。随着医学与基因科技的发展,治疗遗传病的药物从无到有,越来越多的生死绝症正在变成有药可医;通过产前诊断和辅助生殖技术,患者也可以生育健康的下一代。
2018 年 5 月,脊髓小脑性共济失调被纳入中国第一批罕见病目录。相信在不远的未来,科技之光终会照耀到 SCA 病友群体。希望患有遗传疾病的朋友们不要对生活失去信心!疾病给了我们痛苦,唯有报之以歌。从绝望中寻找希望,在黑暗中摸索光明,我想这正是我们要做的事情。
过去已往,未来可期!
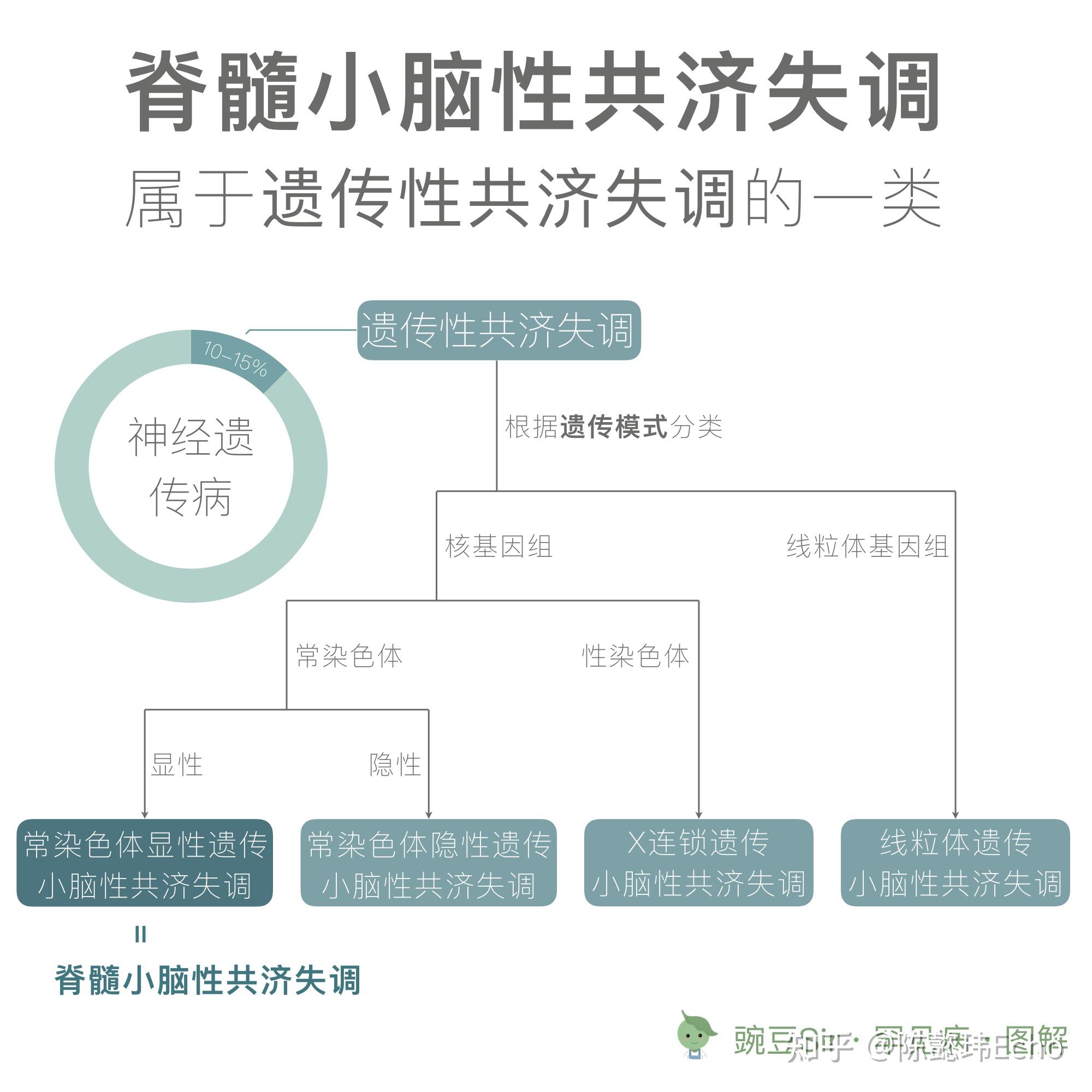
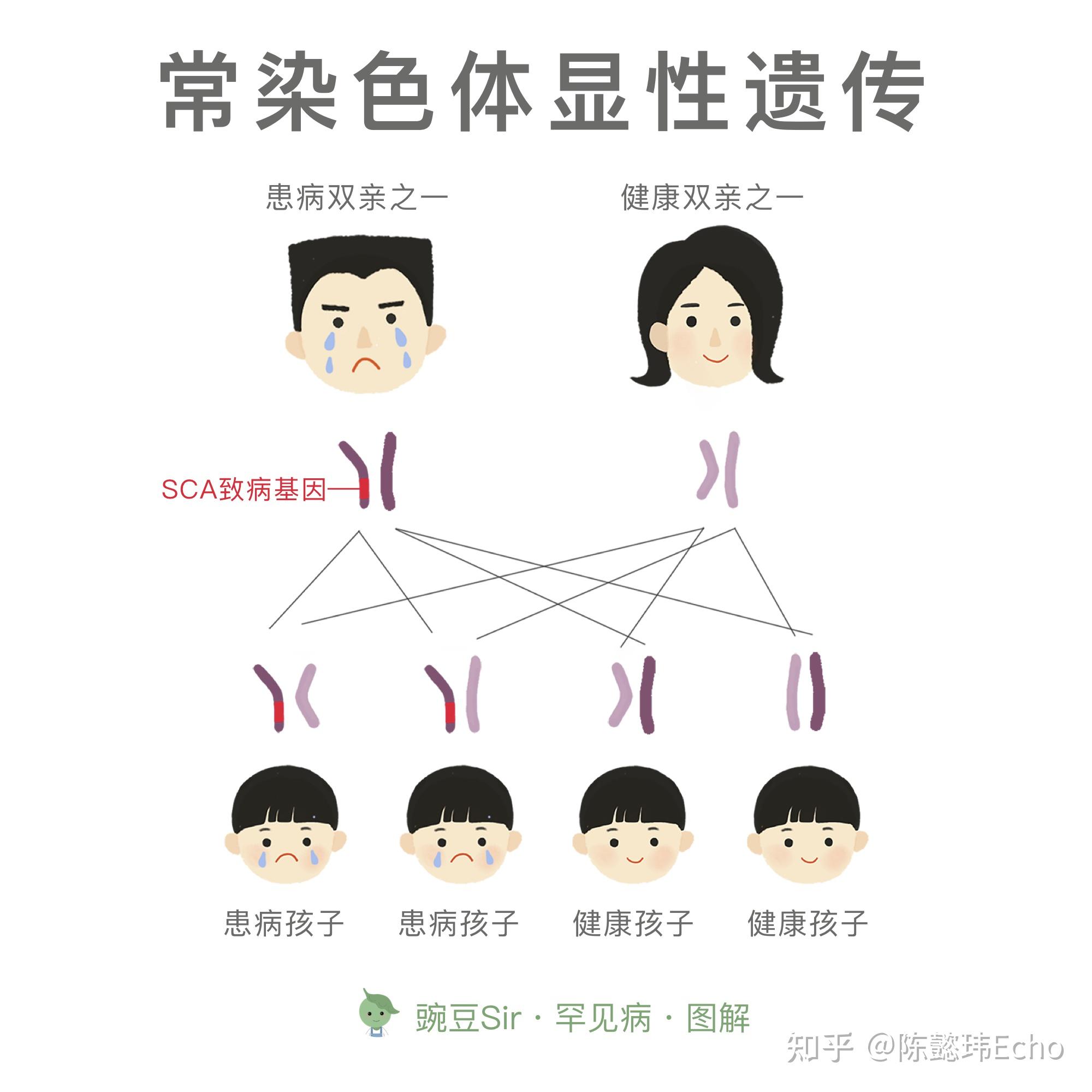
<hr/>脊髓小脑性共济失调 Spinocerebellar ataxia (SCA)
一组由基因变异引起的以脊髓和小脑损害为主要临床表现的常染色体显性遗传的神经退行性疾病。
别名
- 常染色体显性小脑性共济失调 Autosomal dominant cerebellar ataxia [1-3]
- 常染色体显性脊髓小脑性共济失调Autosomal dominant spinocerebellar ataxia [1-3]
数据库编号
- DOID:1441 [2]
- ORPHA:99 [3]
- 中国第一批罕见病目录:111 [4]
参考来源 [1]
临床特征
患病率 由于 SCA 是一组疾病,且在世界各地的患病率存在群体/地域差异,因此难以估算准确的全球患病率。根据目前有限的流行病学研究,患病率可能介于 1-5/10 万之间 [1]。
发病年龄 跨度较大,从幼儿期至中年期,多在青年期隐匿起病,症状缓慢进行性加重。动态突变型 SCA 家系常表现出遗传早现,即发病年龄逐代提前 [5]。
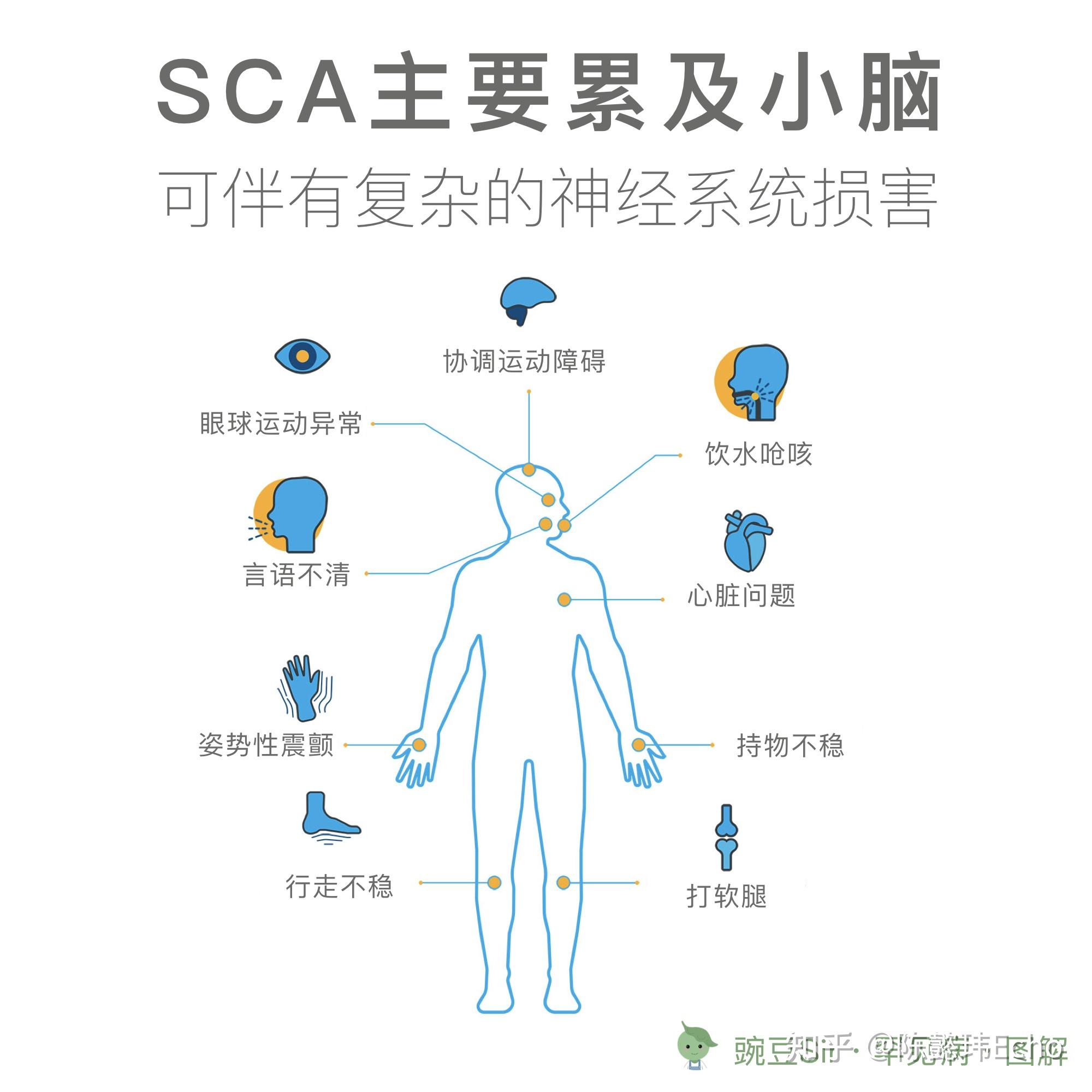
临床表现
- 行走不稳
- 持物不稳
- 言语不清
- 眼球运动异常
- 姿势性震颤
- 饮水呛咳
参考来源 [6]
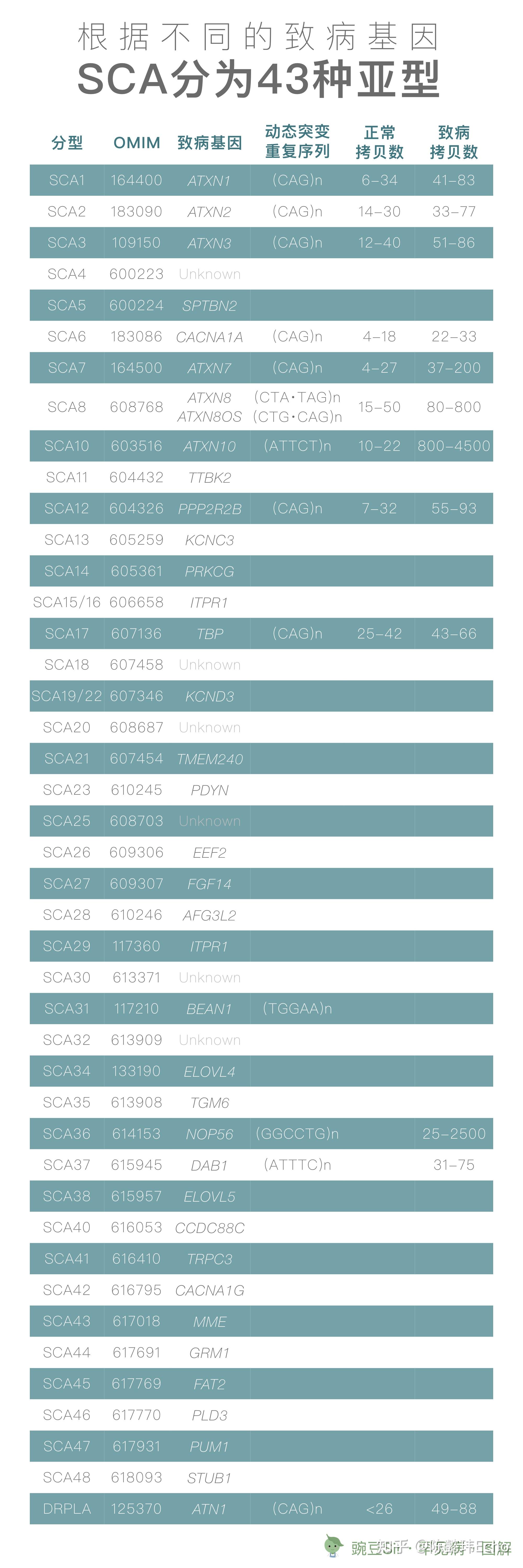
基因分型
SCA 是一组遗传异质性很高的疾病。截止 2020 年 2 月,已发现 43 个导致 SCA 的致病基因区间,其中 38 个致病基因被克隆。根据不同的致病基因,SCA 可分为 43 个亚型 [7-9]。
近年来国内多家三甲医院通过基因学分析,均确认 SCA3 为 SCA 中最常见的类型,约占遗传性共济失调的 40%-60%,其次是 SCA2、SCA1、SCA7、SCA6 和 SCA12 等 [10]。
参考来源 [7-9]
致病机制
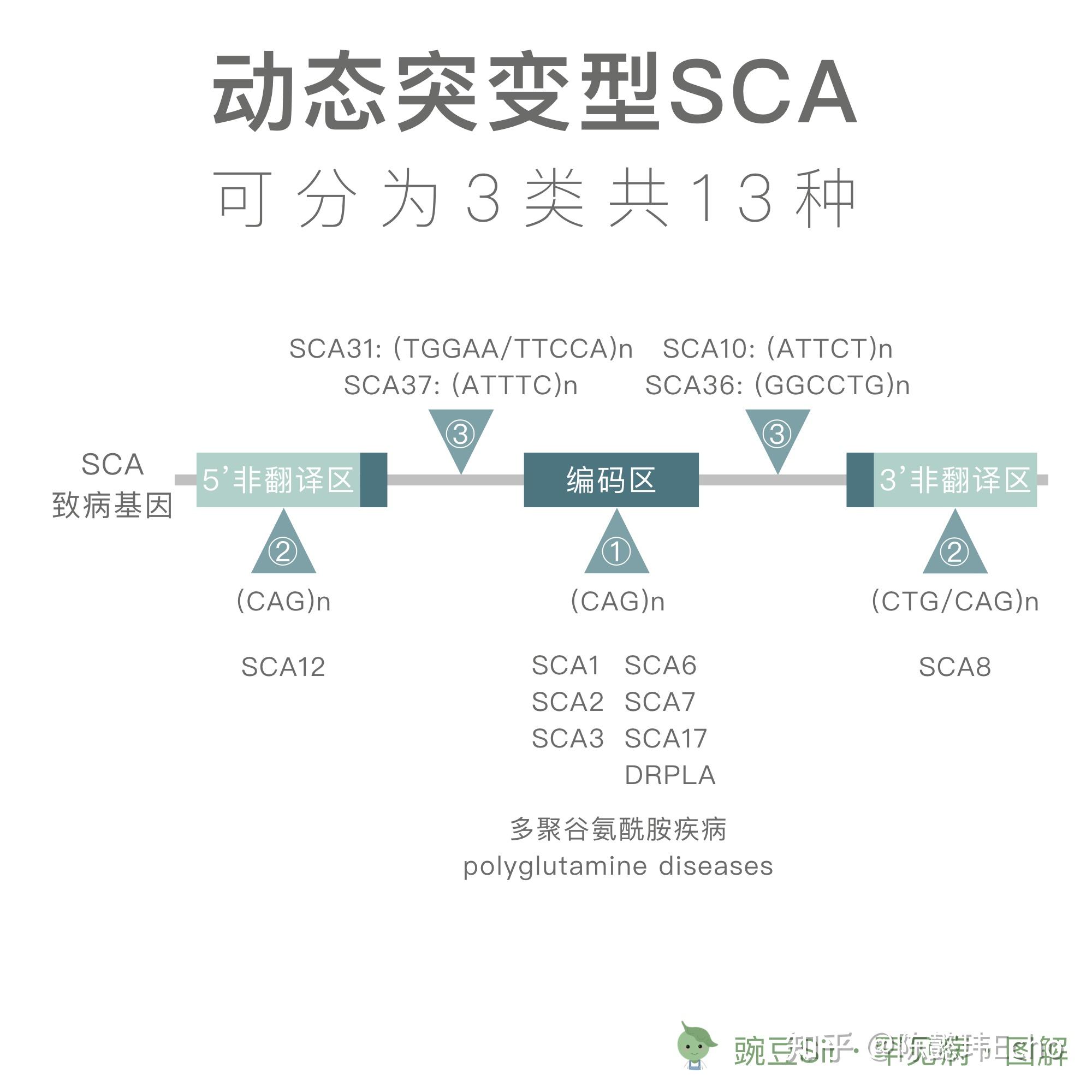
SCA 根据致病变异可大致分为 2 种类型:
1. 动态突变型
① 由于致病基因编码区内的 (CAG)n 三核苷酸重复的异常扩展而致病。三核苷酸 CAG 编码多聚谷氨酰胺 (polyglutamine, PolyQ),因此这类 SCA 也被称为多聚谷氨酰胺疾病;
② 由于致病基因非翻译区内的 (CAG)n 三核苷酸重复的异常扩展而致病;
③ 由于致病基因非编码区内的其他多核苷酸重复的异常扩展而致病。
2. 其他突变型,如点突变、插入/缺失突变
参考来源 [8]
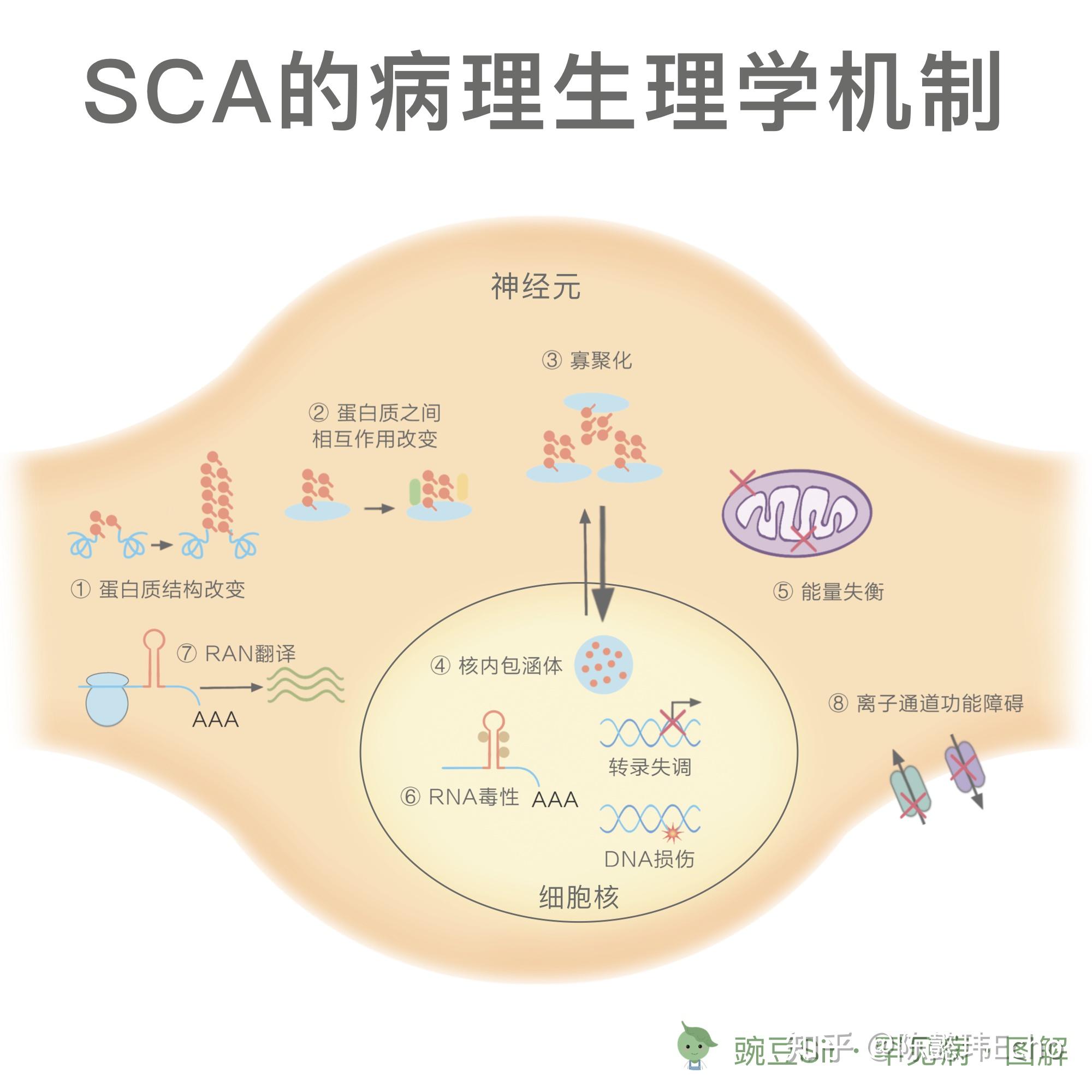
SCA的发病机制尚未完全阐明。目前,对于特定SCA的研究发现了一些可能引起神经元退化的病理生理学机制:
1. 多聚谷氨酰胺链的异常扩展: SCA1, 2, 3, 6, 7, 17, DRPLA
① 改变蛋白质原本的结构与功能;
② 影响与其他蛋白质的相互作用;
③ 倾向于寡聚化,产生蛋白毒性;
④ 在细胞核内,形成包涵体,引起基因转录失调、DNA 损伤,破坏核完整性;
⑤ 在细胞质中,引起能量失衡,最终造成整个神经元细胞的损伤甚至死亡。
2. 非编码区的重复序列异常扩展: SCA8, 12, 10, 31, 36, 37
⑥ 在细胞核内,与关键的 RNA 结合蛋白螯合,干扰正常剪接,产生 RNA 毒性;
⑦ 在细胞质中,引发重复序列相关的非 ATG 介导的翻译,生成易于聚集的多肽,产生蛋白毒性。
3. 离子通道基因变异: SCA6, 13, 19/22, 15/16, 29, 41, 42, 44
⑧ 导致通道功能障碍,引起小脑回路紊乱,造成共济失调。
参考来源 [8]
诊断
① 运动障碍表现 缓慢发生、进展性、对称性共济失调
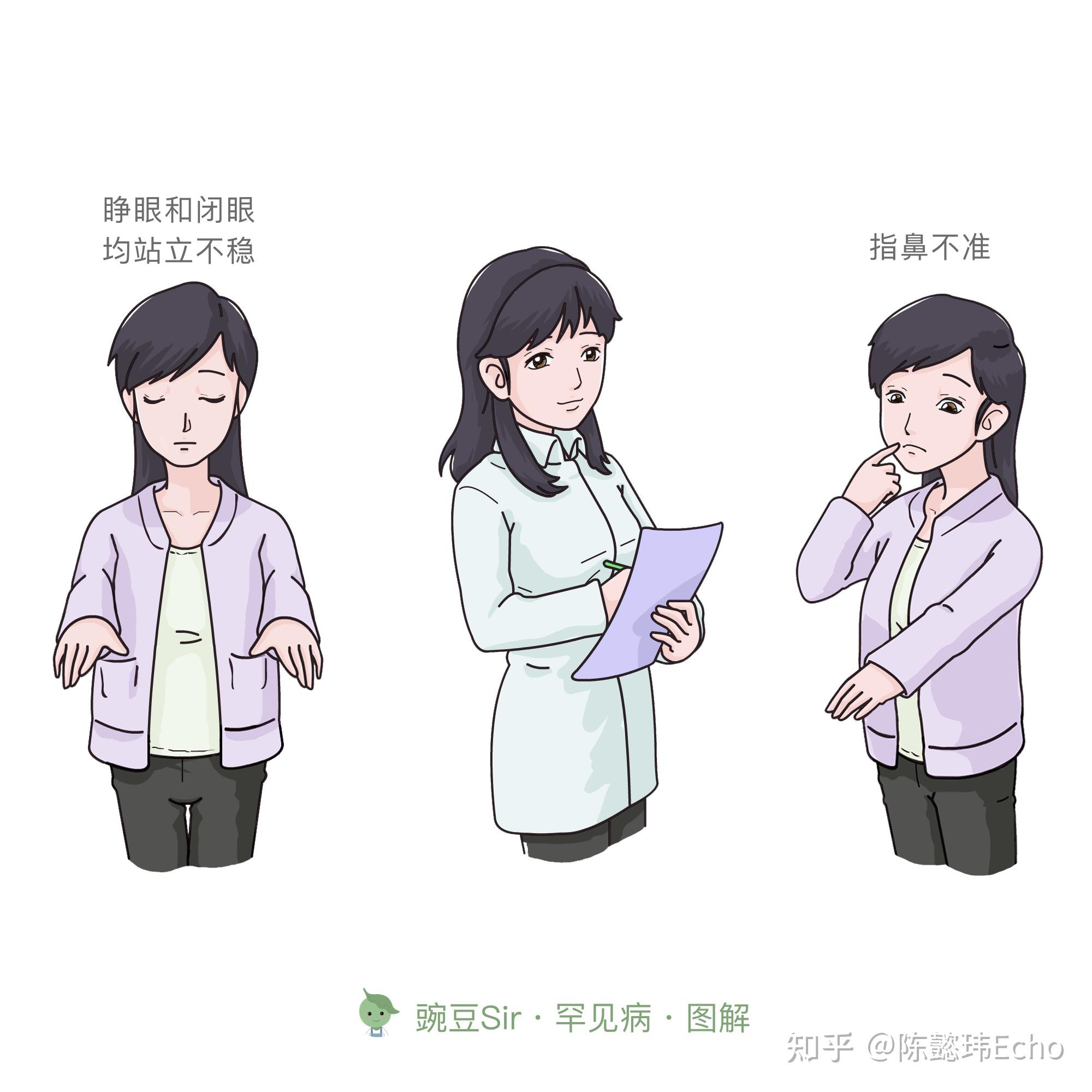
② 家族史 具有常染色体显性遗传特点

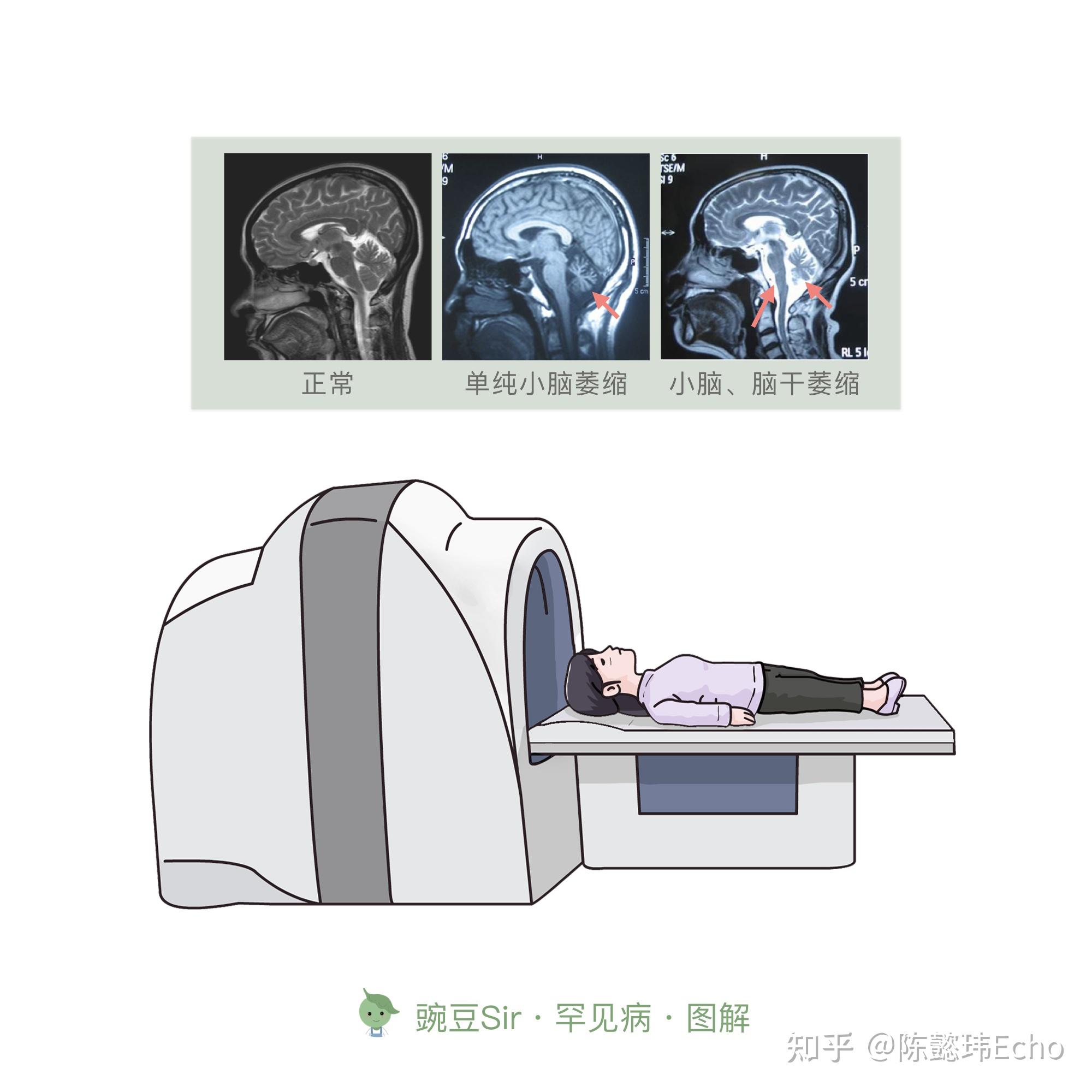
③ 影像学检查 小脑或脑干不同程度萎缩
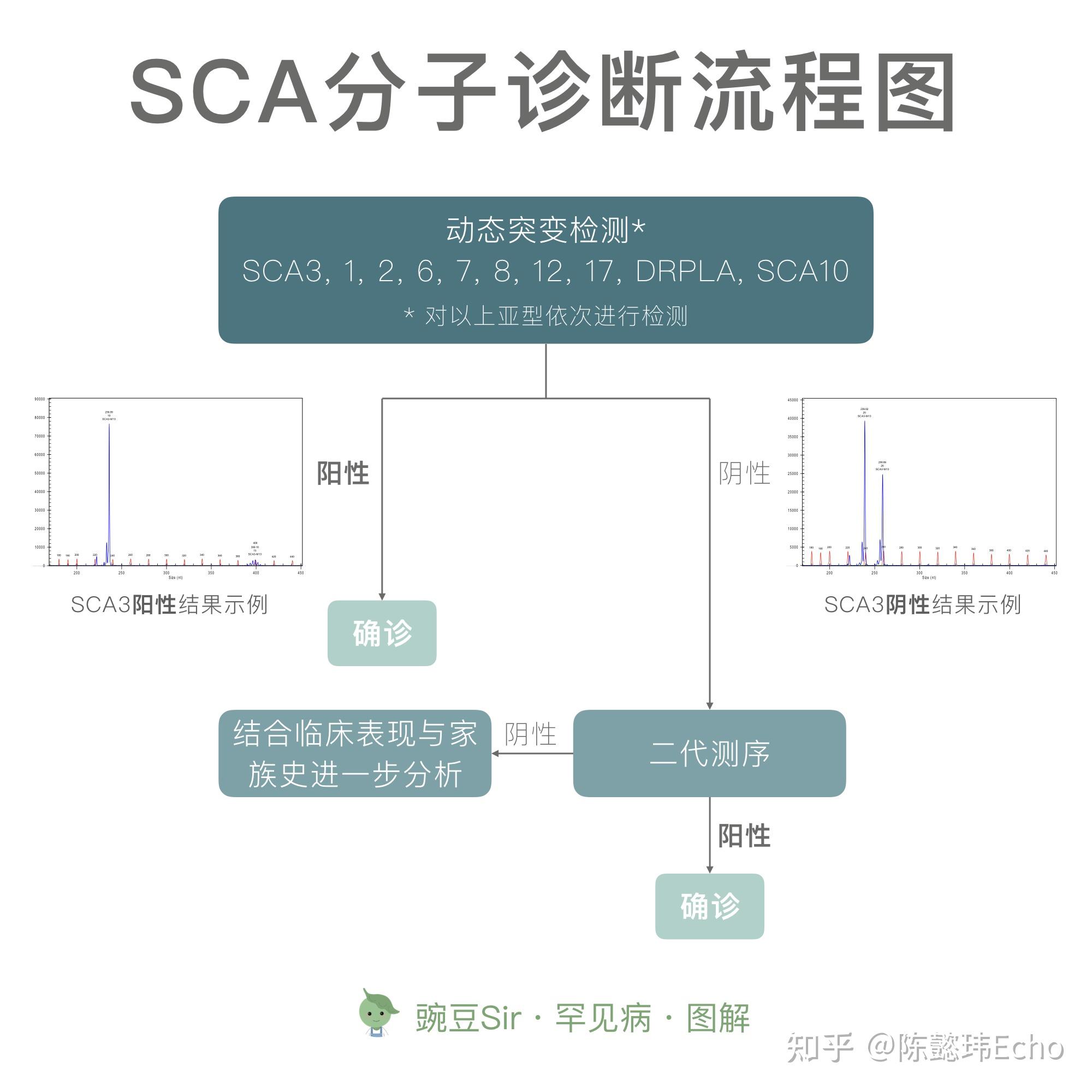
④ 分子诊断 检测SCA致病基因
治疗
原则 目前尚无能够完全阻止病情进展的方案或有效的病因治疗,临床上以对症和支持治疗为主,主要目标是减轻症状、延缓病情进展,改善日常生活自理能力。
对症药物治疗
① 痉挛疼痛 可用巴氯芬、盐酸替扎尼丁。
② 帕金森病样表现和肌张力障碍 左旋多巴、盐酸苯海索(安坦)、金刚烷胺、氯硝西泮有助于症状改善。
③ 睡眠障碍 氯硝西泮可改善症状。
④ 情感障碍 须予重视,并及时给予抗焦虑、抗抑郁药物治疗。
非药物治疗
① 康复训练 帮助患者提高生活自理能力,推迟和减轻致残的程度和时间。
② 言语训练 改善语言功能,帮助患者更好地沟通。
③ 作业治疗 帮助患者使用辅助书写、进食、自我照料与行动的一系列用具。
④ 心理干预 增强患者对疾病的认知与自信心。
遗传咨询
建议生育过患者的家庭的成员,在婚前或孕前到有资质的医疗机构接受遗传咨询,筛查是否携带与患者相同的致病基因。
根据 SCA 常染色体显性的遗传方式,结合基因检测的结果,可计算后代的遗传风险。如已怀孕,可对胎儿进行产前诊断,诊断结果可作为是否继续妊娠的重要参考;也可应用试管婴儿体外基因诊断技术选择正常的胚胎后再植入母体子宫继续妊娠。
社会组织
北京企鹅之家小脑萎缩症病患关爱中心(原中国共济失调病友协会)于 2010 年 9 月 28 日由 SCA 患者和家属共同发起成立,并于 2018 年 8 月 28 日在北京市民政局注册,致力于
- 为 SCA 患者提供最新的研究进展信息;
- 帮助 SCA 群体在医疗、生活、教育、就业等方面获得平等权益;
- 推动保障罕见病群体合法权益相关制度、政策的完善。
官网 www.webataxia.com
微信 BJ-CAA(公众号)
SCA Global 是一个灵活开放的研究平台,旨在
- 更好地理解各种 SCA 的表现、演变和影响;
- 开发并验证可用于未来干预试验的生物标志物;
- 招募愿意参加临床试验的 SCA 患者。
官网 ataxia-global-initiatives.net/sca-global
National Ataxia Foundation 于 1957 年由共济失调患者发起成立,旨在
- 支持共济失调的科研活动;
- 为患者家庭提供服务;
- 寻找治愈的方法。
官网 ataxia.org
Euro-ataxia 是一个国际性的非营利组织,致力于在欧洲范围内
- 推动共济失调的研究和治疗;
- 在临床医生,科学家和社会中提高对共济失调的认知;
- 鼓励成员间就最新研究自由交流信息;
- 促进国家共济失调组织之间的国际合作;
- 对与共济失调患者福利相关的社会、政治和文化事务进行调查,并促进和改善信息交换。
官网 www.euroataxia.org
Ataxia UK 是英国领先的全国性慈善机构,致力于为任何类型的共济失调患者提供服务,包括:
- 为探索治疗方案的研究项目提供资金;
- 为患者家庭建议,信息和支持。
官网 www.ataxia.org.uk
<hr/>专家的话
顾卫红 医生
我从 1997 年开始,和王国相教授一起做 SCA 家系调查,至今已过去 20 多年,在临床工作中接诊了大量 SCA 患者。我曾经深入走进病友群体,了解他们的生活和想法,有很多感触。
SCA 群体比较大,包括 30 种以上的疾病,大多为中青年期起病。患者具有一定的教育和工作背景,正值人生上升期,发病对于患者和家庭的影响很大,绝大多数患者都会经历逐步接受现实的过程,十分艰难,非常需要心理支持和病友社群的互助,也很需要社会各界的理解与支持。
SCA 病友群的组织和引导非常重要,减少盲目就医,避免上当受骗。提倡科学理性的抗病方法和理念,与疾病共存,积极康复,采用适当的方法进行运动功能训练和呼吸功能训练,同时要避免受伤。
SCA 的确诊需要做基因检测,一半以上的 SCA 家系为动态突变疾病,在做基因检测时首先需要检测动态突变类型。关于 SCA 家系患者后代基因检测的问题,需注意以下三点:
(1)为了避免引起 “知情负担”,不建议未成年、未发病后代检测 SCA 致病基因。
(2)基因检测报告是非常重要的文件,检测机构(医院或者检测公司)须出具正式纸质报告,对于整个家系的病因诊断和遗传咨询都是至关重要的。
(3)SCA 家系患者的后代达到婚育年龄时,在本人知情同意的前提下,在孕前检测是否携带家族的致病突变,如果携带致病突变建议做生殖干预,如果选择做产前诊断,尽量在孕前联系好具有相应资质和检测能力的医疗机构,产前诊断需要按照严格的流程操作,如果意外怀孕,一定要尽早到具有产前诊断资质的医院就诊(不要晚于 10 周)。
<hr/>封面 + 肖像 | 君君
案例 | 君来
科普 | Echo
插图 | 蓝剑儿、翟进、君君
特别感谢香港中文大学生命科学学院陈浩然教授对本文的审阅与指导。
参考文献:
- Bird. “Hereditary Ataxia Overview.” GeneReviews® [Internet]., U.S. National Library of Medicine, 25 July 2019, http://www.ncbi.nlm.nih.gov/books/NBK1138.
- Disease Ontology, Institute for Genome Sciences @ University of Maryland, www.disease-ontology.org.
- Orphanet, INSERM, www.orpha.net.
- 国家卫生健康委员会 等. “关于公布第一批罕见病目录的通知.” 中华人民共和国卫生健康委员会, 2018年6月8日, http://www.nhc.gov.cn/yzygj/s7659/201806/393a9a37f39c4b458d6e830f40a4bb99.shtml.
- 陈静 主编. 罕见疾病. 上海交通大学出版社, 2019. 297-301.
- “What Is Ataxia?” National Ataxia Foundation, http://ataxia.org/what-is-ataxia.
- Online Mendelian Inheritance in Man (OMIM), www.omim.org.
- “HEREDITARY ATAXIAS: DOMINANT.” NEUROMUSCULAR DISEASE CENTER, Washington University, http://neuromuscular.wustl.edu/ataxia/domatax.html.
- Klockgethe et al. Spinocerebellar ataxia. Nat Rev Dis Primers 5, 24 (2019), http://doi.org/10.1038/s41572-019-0074-3.
- 王维治 主编. 神经病学(第2版). 人民卫生出版社, 2013年. 1833-1837.
专栏简介
罕见病,是一类患病率很低的疾病的总称。因为症状复杂但病例稀少,诊断的难度很大。即便在美国,每位罕见病患者平均需要 7.6 年经历 8 位医生,被误诊过 2、3 次才能确诊。在中国,由于医疗资源的缺乏与不均,罕见病专业医生比罕见病更罕见。
自今日起,罕见病科普公益项目“豌豆Sir”将推出《罕见病 · 医者仁心 》人文科普专栏。每期与一位国内的罕见病临床专家合作,以可视化的形式介绍一种罕见病,以医学叙事的方式呈现罕见病患者的典型病程与生活经历。
《罕见病 · 医者仁心 》专栏由中国国家罕见病注册系统*和神州数码医疗科技股份有限公司共同参与策划并启动,旨在为广大一线的医务工作者提供全面、可靠、深入的罕见病知识,以及可实操的诊疗经验。让罕见病不再罕见!
*中国国家罕见病注册系统(National Rare Diseases Registry System of China, NRDRS)作为中国罕见病事业发展的基础建设,依托“十三五”国家重点研发计划精准医学专项“罕见病临床队列研究”,由北京协和医院牵头,汇聚国内 20 家具有罕见病临床研究基础的优势单位共同攻关,自 2016 年 12 月启动注册研究队列至今,截至 2021 年 1 月 17 日,在 185 个疾病分类表单共计注册病例 62780 例。自 2019 年 9 月启动 NRDRS 整体升级,2020 年 5 月更新为 NRDRS V2.0,一直在持续优化系统功能,尽全力为罕见病专业 PI 提供优质注册平台。
官网 中国国家罕见病注册系统 |
|



 发表于 2022-12-9 15:49:20
发表于 2022-12-9 15:49:20