|
|
现状:
当前很多多发转移的胸腺瘤病友在告别了手术、放疗且化疗无效后使用了基于血管生成抑制类的靶向药物进行治疗,治疗效果据文献统计有效率接近半成左右,个人了解多数病友都有很不错的抑制效果,但也不乏有因为严重的腹泻、出血等副作用不可耐受不得不中途停药的无奈之举,剩下的病友也面临着平均耐药期一年左右的窘迫情况。通过查阅相关文献和案例发现近几年大夫、病友有些主推或试水多种靶向药联合或免疫+靶向的方法路子,也取得了一些效果,对于C型病友可能两种方案(免疫+VEGFR2、VEGFR2+EGFR)都适用,而B型病友则要考虑免疫反应的反噬而只能先试后者。无论如何这终归是一条路子,在使用中因为两种药物的叠加导致副作用也会有更大的加重,进一步加剧了身体的不可耐受,同时多药混用也要考虑药物相互作用,配伍是否有禁忌情况?此篇论文的研发发现就是融合双靶点的新药,若后期成功上市可解决了副作用叠加的同时又进一步提高了靶向作用,更好的控制了肿瘤生长。
血管内皮生长因子受体 (VEGFRs) 是受体蛋白酪氨酸激酶家族,在调节肿瘤诱导的血管生成中起重要作用。目前,VEGFR抑制剂已广泛应用于各种肿瘤的治疗。然而,目前的VEGFR抑制剂由于临床疗效有限和潜在的毒性在一定程度上受到限制,阻碍了其临床应用。因此,需要开发新的策略来改善临床结果并最大限度地减少 VEGFR 抑制剂的毒性作用。鉴于 VEGFR 和其他疗法在肿瘤发展和进展中的协同作用,VEGFR 双靶点抑制剂因其良好的药效学、低毒性和抗耐药作用而成为一种有吸引力的方法。
介绍
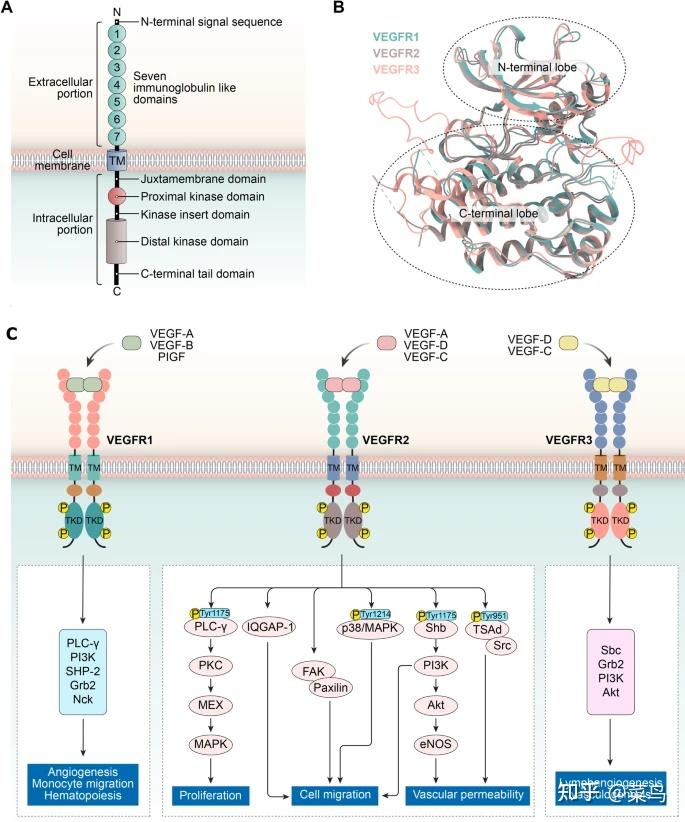
异常的血管生成可以被认为是肿瘤进展和转移的必要先决条件。现有证据表明,许多细胞外、细胞表面和细胞内分子可以直接或间接调节血管生成 [ 1 , 2 ]。特别是血管内皮生长因子(VEGFs)及其与膜受体的相互作用在血管生成过程中具有重要意义。在哺乳动物中,VEGF 亚型 [VEGF-A、B、C、D 和胎盘生长因子 (PIGF)] 由 VEGF 相关基因编码,并与 VEGFR-1/Flt-1 的 VEGF 受体 (VEGFRs) 家族特异性相互作用, VEGFR2/KDR 和 VEGFR-3/Flt-4 [ 3 , 4]。这些受体具有高度的结构相似性,但在激活方式、信号转导和生物学功能方面存在差异 [ 5 ]。表1总结了这些受体的特定配体、主要功能和不同的结构域。简而言之,VEGFR1、VEGFR2 和 VEGFR3 分别对造血细胞、血管内皮细胞和淋巴管内皮细胞的发育至关重要。然而,VEGFR3 及其配体在淋巴管生成和肿瘤细胞扩散到区域淋巴结中起关键作用[ 6、7]。在结构上,VEGFRs 由一个细胞外部分组成,该部分由一个细胞外配体结合结构域 (ECD) 和七个免疫球蛋白样结构域 (IgD)、一个跨膜结构域 (TM)、一个近膜结构域 (JMD)、一个酪氨酸激酶结构域 (TKD) 组成。 ) 插入约 80 个残基和一个羧基末端(图 1 A、B)[ 8 ]。VEGFR的激活可以通过配体结合来介导。随后,配体诱导的 VEGFR 胞内结构域构象变化促进受体二聚化,导致特定酪氨酸残基的自磷酸化和几种下游酶通路的激活,包括 p38/MAPK、RAS/RAF/MEK/ERK 和 PI3K/AKT/ mTOR(图 1C)。同时,一些受体经历内化并形成内体。在内化的早期,受体仍然作为内体的膜蛋白成分存在,而这个由微管和液泡组成的受体区室广泛分布于细胞质中。在受体穿过内体膜的过程中,配体-受体复合物保持完整,受体的激酶功能保持激活。最后,含有配体-受体复合物的膜片段作为小囊泡被挤入内体的内腔,从而形成多泡内体。结果,质膜上的配体-受体复合物到达内体的管腔,并与管腔的其他内容物一起广泛分布在整个细胞质中。9 ]。重要的是,先前已经表明 VEGF 可以诱导 VEGFR1 和 VEGFR2 的内化 [ 10 ]。
表 1 VEGFR 家族的独特特征
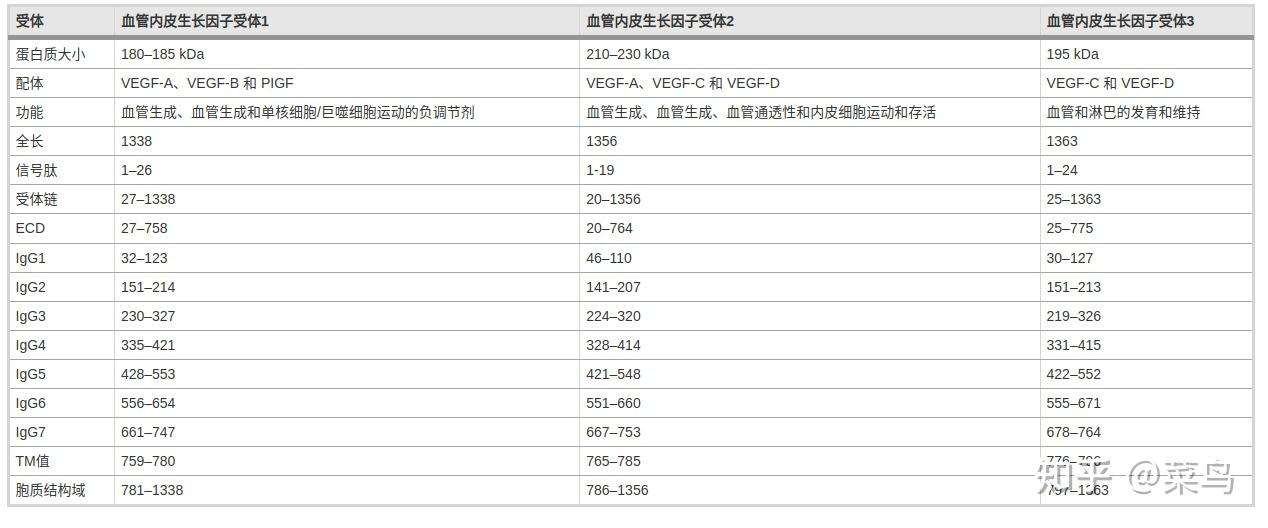
图一
VEGFR蛋白结构域结构示意图;BVEGFR3(粉红色)、VEGFR2(PDB ID:3WZE,棕色)和 VEGFR1(PDB ID:3HNG,蓝色)的晶体结构重叠。利用VEGFR1和VEGFR2的结构构建人VEGFR3的同源模型;VEGFR信号调控肿瘤血管生成的C机制
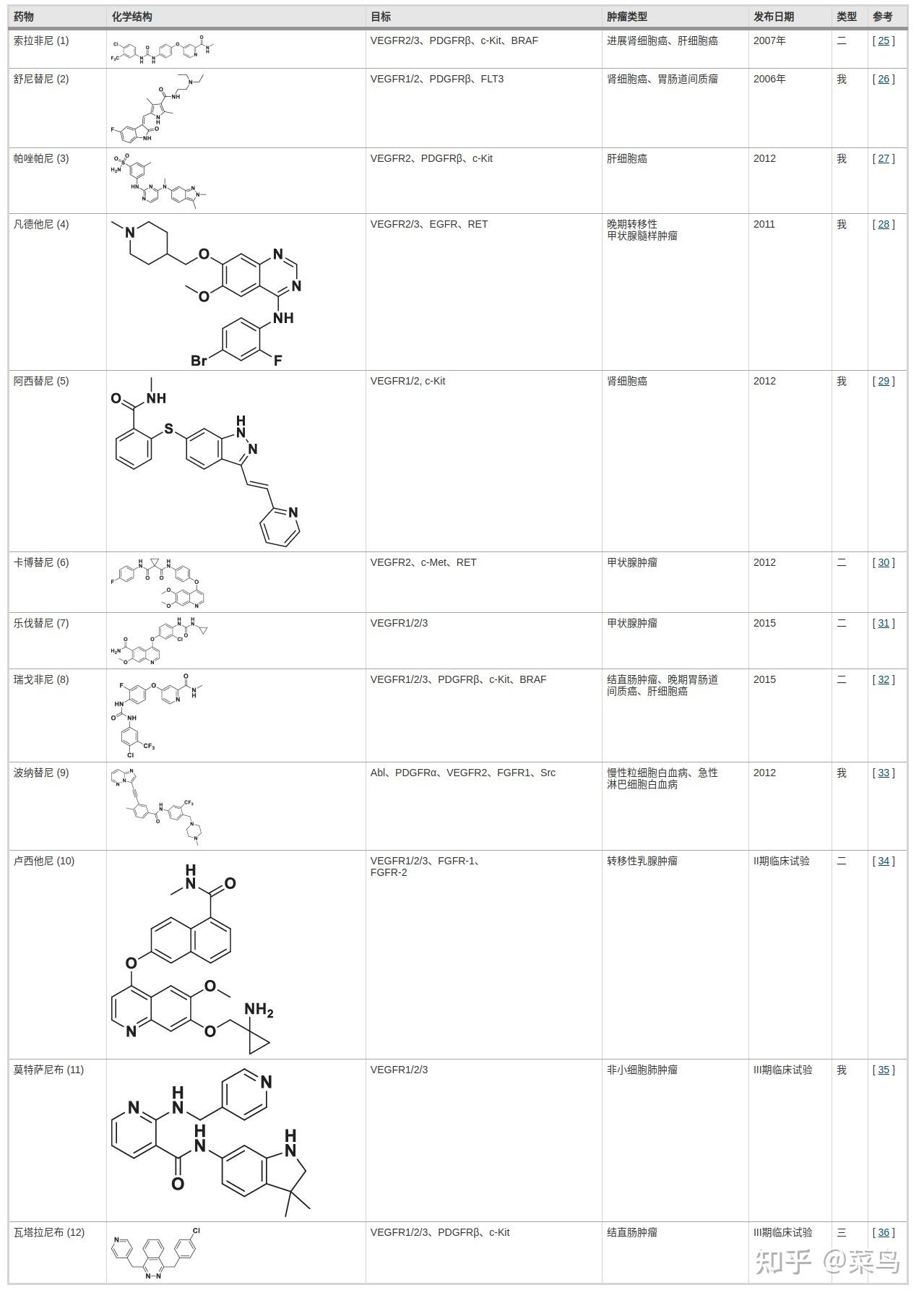
功能失调的 VEGF-VEGFR 信号轴广泛涉及人类疾病,尤其是肿瘤。靶向 VEGF 信号的抑制剂,包括靶向 VEGF 的单克隆抗体和靶向 VEGFR 的小分子,已显示出对不同类型实体瘤的治疗效果。具体而言,贝伐单抗作为一种靶向VEGF的重组人源化单克隆抗体,发挥着有益的临床效果。然而,抗VEGF单克隆抗体的主要问题是免疫原性高、成本高、稳定性低。此外,抗 VEGF 单克隆抗体的临床应用受到与抑制生理性血管生成相关的相当大的副作用的严重限制,生理性血管生成是抗血管生成疗法最常见的副作用之一。目前,11]。迄今为止,已从www.ClinicalTrials.gov网站检索到超过 340 项与 VEGFR 抑制剂相关的临床试验。具体而言,几种VEGFR抑制剂已获批临床使用,其疗效结果总结于表2。值得注意的是,VEGFR 抑制剂可分为三类:I 型抑制剂、II 型抑制剂和 III 型抑制剂 [12]。I 型抑制剂 [例如,舒尼替尼 (2)、帕唑帕尼 (3)、凡德他尼 (4)、阿西替尼 (5)、波纳替尼 (9) 和莫特萨尼 (11 ))],也称为ATP竞争性抑制剂,可与腺嘌呤区域产生疏水相互作用,并与受体活性位点周围的残基形成1-3个氢键,从而竞争结合活性“DFG-in”构象在 ATP 结合口袋中 [13]。II 型抑制剂 [例如,索拉非尼 (1)、卡博替尼 (6)、乐伐替尼 (7)、瑞戈非尼 (8) 和卢西他尼 (10)] 的特征在于与激酶的非活性“DFG-out”构象结合并占据与 ATP 结合位点相邻的疏水袋 [14,15]。III 型抑制剂 [例如,伐他拉尼 (12)],或称为共价抑制剂,可以通过在激酶的特定位点不可逆地结合半胱氨酸来发挥其药理功能 [16]。到目前为止,许多批准的 VEGFR 抑制剂是 I 型抑制剂,其靶向 ATP 结合口袋。基于 VEGFR2 的 X 射线晶体结构,据报道几种 I 型抑制剂对肿瘤抑制具有调节作用。然而,一些研究表明,II 型抑制剂比 I 型抑制剂具有某些优势,包括提高效力和选择性 [17,18]。在结构上,向不太保守的变构疏水后袋的延伸促进了 II 型抑制剂的亲和力和选择性 [19]。此外,共价酶抑制剂已被广泛用作治疗剂[20]。一般来说,这些抑制剂大多可以达到持续改善,甚至治愈一些肿瘤患者。然而,它们的临床应用受到治疗耐药性、疗效有限和脱靶毒性的限制 [6]。首先,对 VEGFR 抑制剂的耐药机制分为以下几部分: (i) 激活替代的促血管生成信号通路;(ii) 募集局部和远端基质细胞;(iii) 肿瘤血管化的替代模式(例如,缺氧)。其次,由于 VEGFR 和其他受体激酶结构域的相似性,这些抑制剂对其他靶标(如 PDGFR、c-KIT 和 FLT3)显示出交叉抑制活性,导致可能的脱靶效应。已经研究了 VEGFR 抑制剂的几种临床毒性作用,例如高血压、蛋白尿、甲状腺功能减退、白质脑病综合征和动脉血栓形成。最后,21、22]。_因此,克服这些缺点的临床策略,如联合治疗,需要得到很好的关注[23,24]。
表 2 临床批准的 VEGFR 抑制剂汇总
人类肿瘤的联合治疗不仅增加了效力,而且减少了潜在的不良事件 [ 37 ]。由于这些酶在肿瘤中的治疗效果和相关生物学功能已被揭示,因此认为它们与 VEGFR 抑制剂(例如,表观遗传剂、免疫治疗药物和其他 RTK 抑制剂)的组合在抗肿瘤治疗中是有希望的。然而,应该谨慎的是,复杂的剂量/时间表、可疑的药代动力学/药效学特征和潜在的不良事件需要深入探索 [ 38 , 39]。作为联合治疗的替代策略,双靶点或多靶点药物的特点是不良药物相互作用 (DDI) 的风险降低,药代动力学 (PK) 曲线更好,安全性得到保证 [ 40 ]。基于这些概念,VEGFR 双靶点抑制剂正在成为一种有吸引力的方法。
鉴于 VEGFR 和其他疗法在肿瘤发展和进展中的协同作用,新型 VEGFR 双靶点抑制剂的鉴定可能为临床实践提供有效策略。从这个角度总结了双靶点VEGFR抑制剂的研究进展,重点关注双靶点VEGFR抑制剂的合理靶点选择、构效关系(SARs)和药理活性。
VEGFR2作为治疗靶点
如上所述,每个 VEGFR 家族都具有独特的特征。其中,VEGFR2已被确定为有前景的肿瘤治疗靶点[ 41 ]。越来越多的证据证实,新生血管肿瘤内皮细胞中VEGFR2的异常表达与多种肿瘤的发生、发展密切相关[ 42 , 43 ]。通过阻断血管生成和淋巴管生成,所有 VEGFR2 抑制剂对不同类型的肿瘤提供不同程度的临床益处,尽管它们中的大多数缺乏特异性 [ 44 ]。
VEGFR2抑制剂的研究现状
在医药领域,高效VEGFR2抑制剂的发现和开发一直是研究热点。VEGFR2 与 FDA 批准的抑制剂复合的共晶体结构揭示了基于结构的 VEGFR2 抑制剂设计的结构信息。在结构上,VEGFR2 的催化结构域作为具有小 N 叶和大 C 叶的双叶结构,在这些抑制剂的抑制效力中起关键作用。具体来说,VEGFR2 的活性位点由以下亚区组成:疏水区 I(由残基 Leu840、Phe918 和 Gly922 包裹)、疏水区 II(由残基 Leu889、Ile892、Val898 和 Ile1044 包裹)和一个接头区(包裹在由残基 Ala866、Val914、Leu1035 和 Cys1045)[ 45 ]。如图所示。 2,在复杂的部分 FDA 批准的抑制剂中 VEGFR2 的共晶结构表明,这些抑制剂虽然在结构上高度多样化,但具有决定性的药效团特征。首先,主要骨架的扁平杂芳环系统通过与Cys残基和催化ATP结合袋中的必需残基形成关键氢键而采用活性位点。因此,至少一个氢键受体应包含在该平面系统中(优选 N 原子,其次是 O 原子)。其次,ATP 结合口袋和酶的 DFG 结构域之间的接头区域被中心芳环或间隔物占据。第三,作为药效团的官能团,例如酰胺或脲,分别与 C 螺旋中 Glu885 残基的侧链和 DFG 基序中 Asp1046 残基的主链 NH 形成两个氢键。第四,这些抑制剂的末端疏水部分与变构疏水袋形成疏水相互作用[46、47 ]。_ 基于这些发现,据报道,几种含有不同核心的 VEGFR2 抑制剂可抑制肿瘤生长。
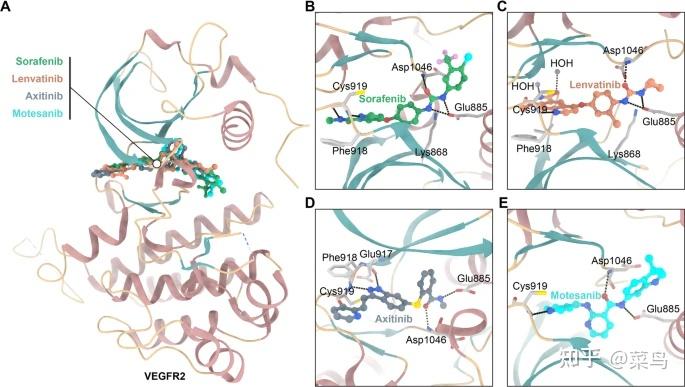
图二
FDA 批准的 VEGFR 抑制剂——VEGFR2 复合物的晶体结构。对应的 PDB 代码为 3WZE(索拉非尼)、3WZD(乐伐替尼)、4AGC(阿西替尼)和 3EFL(莫特塞尼)。氢结合(黑色)相互作用显示为虚线。AVEGFR2与索拉非尼(绿色)、乐伐替尼(橙色)、阿西替尼(灰色)和莫特塞尼(蓝色)的共晶结构重叠;B-EVEGFR2 与索拉非尼、乐伐替尼、阿西替尼和莫特塞尼的结合模式
VEGFR2抑制剂和其他抗肿瘤药物的协同作用
通过临床前或临床评估,VEGFR2抑制剂联合其他抗肿瘤药物的抗肿瘤效力已被广泛鉴定。现有证据表明,其他 RTK 抑制剂和 VEGFR 抑制剂的组合发挥了有利的临床前景 [ 48 , 49]。首先,表皮生长因子受体(EGFR)双重抑制剂西妥昔单抗和 VEGFR 抑制剂索拉非尼的联合治疗在 II 期临床试验(NCT00326495)中显着增强了 KRAS 突变的转移性结直肠肿瘤的临床获益。其次,据报道,VEGFR 的异常表达和成纤维细胞生长因子受体 (FGFR) 的遗传改变与实体瘤的发展有关,协同促进血管生成和纤维化。一些临床证据表明,卢西他尼作为一种双重 VEGFR-FGFR 抑制剂,对实体瘤表现出显着的抑制活性 (NCT01283945)。最后,50 ]。临床证据还表明,c-Met 抑制剂 tivantinib 与 VEGFR 抑制剂 pazopanib 联合用于晚期实体瘤(NCT01468922)具有增强的治疗效果。
快速加速纤维肉瘤 (RAF) 同源物,作为丝氨酸苏氨酸激酶,在调节 RAS-RAF-MEK-ERK 通路中具有重要意义,该通路已被强调为有效的抗肿瘤靶点。在哺乳动物基因中,RAF 同源物由三个独立的基因编码,包括 ARAF、BRAF 和 CRAF。其中,BRAF 表现出最显着的反应性,可以通过肿瘤细胞的突变激活。迄今为止,已在不同类型的肿瘤中检测到缬氨酸 600 突变(V600D、V600E、V600K 和 V600R)。与野生型 BRAF 相比,最常见的突变 BRAF V600E可以显着提高(约 600 倍)激酶活性并最终促进肿瘤的发展 [ 51 , 52]。最近,多项研究表明,BRAF 和 VEGFR2 对肿瘤的发展发挥协同作用,因此,涉及 VEGFR2 抑制剂和 BRAF 抑制剂的联合治疗已被确定为治疗肿瘤的有前景的策略 [ 53 , 54 ]。尽管存在多靶点抑制剂1和8,但 RAF265 是一种有效的 RAF/VEGFR2 双重抑制剂,已被鉴定并成功应用于临床治疗 (NCT00304525) [ 55 ]。
HDAC同工酶可用作肿瘤的有希望的治疗靶标。到目前为止,积累的临床前和临床证据表明,HDAC抑制剂与VEGFR抑制剂的组合在抗肿瘤治疗中是有希望的。特别是,体外和体内证据证明了化合物3和多种 HDAC 抑制剂在逆转耐药性和增强抗肿瘤功效方面的协同作用 [ 56 , 57 ]。此外,一项 I 期试验评估了 HDAC 抑制剂 SAHA 与化合物3联合在TP53 突变患者中的应用,特别是在转移性肉瘤或转移性结直肠肿瘤患者中的应用,并产生了相当大的毒性。58 ]。在另一项 I 期试验中,化合物3和 HDAC 抑制剂 abexinostat 的联合治疗表明,HDAC 抑制可以促进肾细胞癌和其他实体瘤恶性肿瘤患者对化合物3的反应和逆转耐药性 [ 59 ]。作为真核细胞中细胞骨架的关键成分,微管在许多细胞功能中发挥着重要作用。由于它们在关键细胞过程中的特定功能,微管已被强调为有效的抗肿瘤靶标 [ 60 ]。在临床试验中,贝伐单抗(抗 VEGF 单克隆抗体)和紫杉醇(微管靶向剂)的组合显着增加了抗肿瘤反应。61 ]。
雌激素受体α (ERα) 被用作乳腺肿瘤治疗的有希望的治疗靶点,而 VEGFR 在乳腺肿瘤的发展中发挥着重要作用。2010 年,Roshani 等人。报道了联合他莫昔芬、选择性雌激素受体调节剂 (SERM) 和布立尼布在人乳腺癌细胞中的治疗效果。体外和体内证据支持涉及 SERM 和 VEGFR2 抑制剂的联合疗法在提高治疗效果以及抑制 SERM 抗性肿瘤的生长方面的作用[ 62 ]。
以前的研究已经证明了缺氧介导的 PIM1 异常表达在抗血管生成药物耐药中的重要作用 [ 63 ]。2018 年,Andrea L 等人。证明 PIM1 激酶抑制剂与抗血管生成药物的组合在治疗实体瘤方面是有希望的 [ 64 ]。体外和体内研究表明,PIM1 抑制剂和 VEGF 靶向剂的协同作用导致增殖减少、肿瘤脉管系统减少和转移减少 [ 65 ]。
总的来说,这些研究证明了基于 VEGFR 抑制剂的联合疗法的显着治疗优势。具体来说,它们不仅具有良好的效力,而且还可以逆转耐药性。然而,药物联合疗法受到复杂的剂量/时间表、可疑的药代动力学/药效学特征和潜在的不良事件的限制。令人鼓舞的是,通过临床研究和表型筛选对这些药物组合协同功效的认识促进了众多靶点的合理组合,以识别双靶点 VEGFR 抑制剂。
双靶点 VEGFR 抑制剂的设计方法
与单靶点药物和药物联合治疗相比,双靶点策略具有几个优势。首先,双靶点药物既保留了联合治疗的大部分优点,又部分克服了联合治疗的缺点。具体来说,由于一个整合分子,双靶点药物不具有或药物-药物相互作用的风险更低、不良反应更低、更容易预测的 PK 曲线、更低的靶向耐药发生率和更高的患者依从性 [ 66 ]。
已发现合理的靶点组合在双靶点药物的临床成功中发挥关键作用,最终促进双靶点 VEGFR 抑制剂的开发 [ 67 ]。迄今为止,已经做出了巨大的努力来识别双靶点药物。一般来说,药物再利用、基于药效团的组合和计算方法等设计策略经常用于双靶点药物发现[ 68 ]。具体来说,药物再利用是将常规药物应用于新的治疗领域,其特点是开发过程更短[ 69]。此外,大多数双靶点 VEGFR 抑制剂是通过基于药效团的方法鉴定的。这种方法的特点是将两种选择性抑制剂的效力整合到一个分子中,并通过连接或合并选定母体抑制剂的关键药效团来进行。药效团连接方法是通过直接连接药效团或通过共轭接头的简单方法。然而,药效团连接的分子也可能具有高分子量、低吸收和较差的 PK 特性。此外,不合适的接头会阻碍配体部分与靶蛋白的相互作用 [ 70]。与混合设计类似,药效团合并策略是一种通过最大化药效团的重叠水平来获得新化学结构的方法,从而获得比母体药物更小的分子量、简单的骨架和更好的物理化学性质[ 71 ]。然而,母体药物结构的任何改变都可能导致生物活性发生重大变化 [ 72]。因此,在设计双靶点 VEGFR 抑制剂之前,确定 VEGFR 和其他靶点的相互药效团很重要。毫无疑问,药物再利用和基于药效团的方法对于发现双靶点药物至关重要。然而,这些策略的应用是基于已知的小分子,从而导致双靶点 VEGFR 抑制剂的结构多样性较差。如今,计算方法已成功应用于识别具有所需活性谱的双靶点药物,包括基于配体/结构的药物设计、计算机筛选和数据挖掘 [ 73]。具体而言,这些方法能够预测已报道配体的新靶点,并且对于识别或优化所需靶标的新配体也具有重要意义。特别是,通过分子对接、药效团研究和结合口袋相似性搜索鉴定了几种含有新型支架的双靶点 VEGFR 抑制剂,显示出更好的肿瘤治疗效果。
VEGFR2 和其他肿瘤相关靶点的双重抑制剂
VEGFR2-EGFR双重抑制剂
越来越多的证据证实,VEGFR2与EGFR密切相关,参与多种肿瘤的发生发展[ 74、75 ]。因此,VEGFR2抑制剂和EGFR抑制剂的组合在抗肿瘤治疗中是有希望的。因此,据报道,几种 VEGFR2-EGFR 双抑制剂对肿瘤抑制发挥了有希望的作用。VEGFR-EGFR双重抑制剂的化学结构、体外效力和优化如图 3所示。
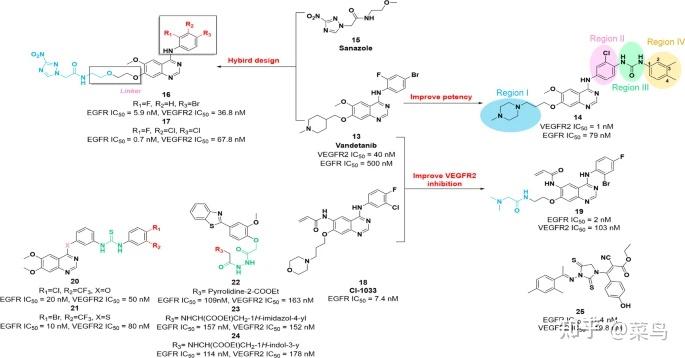
图三
VEGFR2-EGFR 双重抑制剂的化学结构14,16,17和19-25及其对 VEGFR2 和 EGFR 的抑制活性
由于其合适的物理化学性质,二芳基脲和酰胺基团已广泛用于 VEGFR2 抑制剂的设计。2017年,通过优化侧链,在化合物13(vandetanib)的中心核内引入氯,得到类似物14(图 3 )。它显示出对 VEGFR2 和 EGFR 的显着效力,IC 50值分别为 1 nM 和 79 nM。与母体化合物13相比,化合物14对 HT-29 和 MCF-7 细胞具有优异的抑制活性(IC 50 = 1.76 μM 和 7.28 μM,分别)。初步 SAR 研究表明 (i) 在 I 区位置含有 4-甲基哌嗪基团的化合物对 VEGFR2 和 EGFR 发挥更高的效力;(ii) 在区域 II 位置引入氯可以促进激酶对 VEGFR2 和 EGFR 的抑制;(iii) 在区域 III 引入二芳基脲基团有利于提高效力;(iv) 在区域 IV 的 C-3 和 C-4 位置带有甲基的苯环可以提高效力。
越来越多的证据证实,缺氧与多种肿瘤的发生发展密切相关。此外,缺氧是治疗抵抗的主要原因,尤其是在放射治疗中。目前,由于在肿瘤进展和耐药性方面的重要性,缺氧驱动途径中的分子被认为是肿瘤的潜在治疗靶点[ 76 ]。基于这些研究,魏等人制备了一系列以3-硝基-1,2,4-三唑为核心的低氧靶向EGFR/VEGFR2双重抑制剂。[ 77 ]。与13相比,这些化合物中的大多数对 EGFR 具有更强的效力,IC 50在低纳摩尔范围内的值。此外,它们还显示出对 VEGFR2 的良好至中等抑制活性,IC 50值在 36.8 nM 和 4.09 μM 之间的浓度范围内。在这些化合物中,化合物16和17(图 3)显示出对VEGFR2和EGFR的最显着的抑制活性。此外,体外和体内证据证明化合物16具有优越的治疗效果、靶点选择性和可接受的耐受性。进一步的 SAR 研究表明,接头的长度可以显着影响目标化合物对 EGFR 的效力,并且体积大和重的卤素取代苯有助于提高对 VEGFR2 的抑制活性。
2018 年,Bang 等人。根据分子13和第二代 EGFR 抑制剂18 (CI-1033) 的结构鉴定了化合物19,这是一种强大的 VEGFR2/EGFR 双重抑制剂 [ 78 ]。体外酶抑制试验表明,19对 VEGFR2 和 EGFR 均具有有效的抑制效力,IC 50值分别为 103 nM 和 2 nM。此外,19对 EGFRT790M 和 EGFRT790M/L858R 突变体显示出显着的抑制活性,IC 50值分别为 11 nM 和 3 nM。2017 年,化合物20和21基于如图1所示,其在体外对VEGFR2(IC 50 = 50 nM和80 nM)和EGFR(IC 50 = 20 nM和10 nM)表现出选择性、细胞活性和强效。在体内模型中,20和21比分子1 [ 79 ]具有竞争性的肿瘤抑制作用。类似地,Eman 等人也鉴定了化合物22、23和24作为1的衍生物。这些分子对 VEGFR2(IC 50 = 163 nM、152 nM 和 178 nM,分别)和 EGFR(IC 50 = 109 nM、157 nM 和 114 nM,分别)。此外,这些化合物在体外对不同类型的肿瘤细胞显示出低微摩尔效力[ 80 ]。2021 年,Mourad 等人。鉴定了一系列含有 2-硫代咪唑啉-4-one 支架的新型 VEGFR2-EGFR 双抑制剂 [ 81 ]。在这些化合物中,与分子1和 EGFR 抑制剂厄洛替尼相比,化合物25对 VEGFR2 和 EGFR 具有有希望的效力(IC 50 = 19.8 nM 和 11.4 nM),并且对人乳腺癌细胞系 MCF-7 的抗肿瘤作用更强. 此外,25促进细胞凋亡和延长 G2/M 期针对 MCF-7 细胞的细胞周期进程。
双 VEGFR2-FGFR 抑制剂
越来越多的证据证实,VEGF和VEGFR的结合以及FGF2和FGFR的结合协同参与血管生成和纤维形成,从而介导肿瘤的发展[ 82 ]。到目前为止,已经报道了几种含有不同核心的 VEGFR 和 FGFR 双重抑制剂可抑制肿瘤生长 [ 83 ]。它们的化学结构、体外和体内效力以及优化如图 4所示。
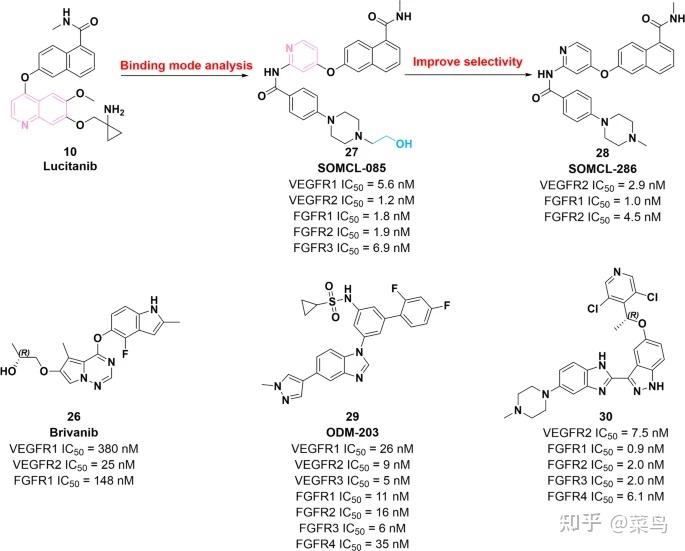
图四 VEGFR2-FGFR 双重抑制剂26 – 30的化学结构及其对 VEGFR2 和 FGFR 的抑制活性
2006 年,化合物26 (brivanib) 被 Bhide 等人鉴定为有前景的 VEGFR2 和 FGFR 抑制剂。[ 84 ]。26对 VEGFR1、VEGFR2 和 FGFR1 具有显着的抑制效力,IC 50值分别为 380 nM、25 nM 和 148 nM。初步 SAR 研究表明 (i) 在吡咯的 5 位引入甲基[2,1- f][1,2,4]三嗪环提高对VEGFR2的抑制活性;(ii)在氟原子的4位取代吲哚环有利于提高对VEGFR2的效力;(iii) 优异的酶效力归因于用醚基团取代了 6 位的酯基团;(iv) CYP3A4可通过引入氨基侧链而受到强烈抑制。然而,26受到口服生物利用度差和吸收低的限制。因此, Cai 等人制备了26的酯类前药。通过在分子26 [ 85 ]的侧链中引入 L-丙氨酸。临床前研究表明,26通过同时阻断 FGF 和 VEGF 通路发挥显着的抗血管生成功效 [ 86 ]。到目前为止,已经有26项临床试验用于治疗不同类型的肿瘤(NCT04395612、NCT03895788、NCT03516071和NCT04212221)。
基于对10和 FGFR 结合模式的研究,进一步发现化合物27 (SOMCL-085) 对 FGFR1、FGFR2、FGFR3、VEGFR1、VEGFR2、PDGFRα 和 PDGFRβ 具有强大的效力(IC 50 = 1.8 nM、1.9 nM、分别为 6.9 nM、5.6 nM、1.2 nM、22.6 nM 和 7.8 nM)。具体而言,将10的喹啉片段打开,引入酰胺作为氢键受体和供体,得到27 。在以下体外和体内试验中,化合物27被确定具有相当大的选择性和抗增殖活性 [ 87 ]。2018 年,魏等人。鉴定化合物28(SOMCL-286) 基于分子27的结构,它是一种有效的 VEGFR2/FGFR 双重抑制剂 [ 88 ]。体外酶抑制试验表明,28对 VEGFR2、FGFR1 和 FGFR2 具有有效的抑制效力,IC 50值分别为 2.9 nM、1.0 nM 和 4.5 nM。然而,与分子27相比, 28对 VEGFR2 和 FGFR 具有更高的选择性。因此,28理论上具有优越的疗效和低毒性。然而,化合物28受到口服生物利用度差的限制,具有 14.9 的低F %。
Compound 29 (ODM-203) 作为一种有效的 VEGFR2/FGFR 抑制剂,对 VEGFR 和 FGFR 家族具有显着的抑制活性,IC 50值在低纳摩尔范围内。此外,它对 VEGFR 和 FGFR 的选择性高于其他激酶。体外和体内证据已证明29在 FGFR 依赖性细胞系 RT4 中具有显着的肿瘤抑制作用 (TGI = 92%) [ 89 ]。值得注意的是,它已在实体瘤治疗的临床试验中进行了评估 (NCT02264418) [ 90 ]。
2016 年,严等人。通过基于知识和结构的方法设计和合成 FGFR 和 VEGFR2 的双重抑制剂 [ 91 ]。其中,分子30含有 3-苯并咪唑-5-吡啶烷氧基-1H-吲哚支架,对 VEGFR2 和 FGFR1-4 显示出显着的抑制活性,IC 50值为 7.5 nM、0.9 nM、2.0 nM、2.0 nM 和 6.1纳米,分别。此外,化合物30不仅在体外有效抑制一组 FGFR 扩增的细胞系,而且在体内也表现出相当大的生物利用度 (33% F) 和肿瘤生长抑制 (TGI = 96.9%)。
双 VEGFR2–c-Met 抑制剂
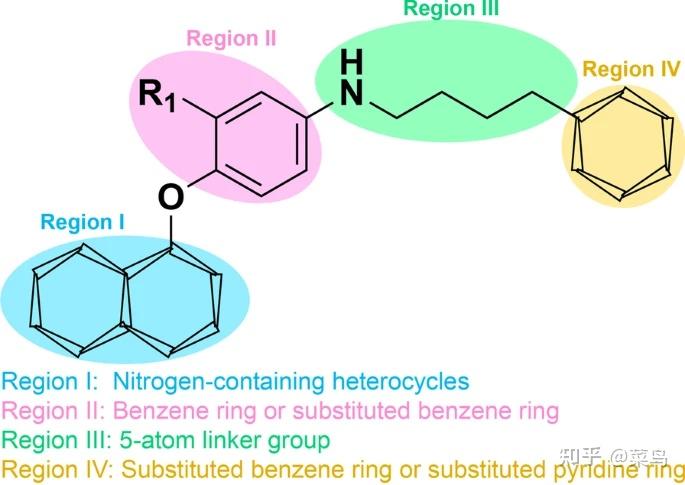
虽然 VEGFR 抑制剂和 c-Met 抑制剂的组合抑制了 VEGFR 和 c-Met 信号通路,但它显着抑制了不同类型肿瘤的发展 [ 92 ]。因此,双靶点 VEGFR/c-Met 抑制剂的鉴定得到了加强。如表3所示,几种基于吡啶或嘧啶的 VEGFR2/c-Met 抑制剂已被鉴定并用于临床试验,包括31(福替尼)、32(戈伐替尼)、33(多维替尼)、34(替沃扎尼) ), 35 (BMS-794833), 36 (BMS-777607), 37 (MGCD-265), 38 (AC480), 39(CP-724714) 和40 (AMG-458)。这些抑制剂的特点是作用于多个靶点并对肿瘤细胞发挥显着的效力。它们的活性支架值得进一步研究,从而促进 VEGFR/c-Met 双重抑制剂的开发。如图 5, 大多数 VEGFR2/c-Met 双重抑制剂具有以下特点: (i) 在 I 区, 可以引入不同的含氮芳香杂环, 包括吡啶、吡咯烷和喹唑啉, 与氨基酸残基形成氢键VEGFR (Cys919) 和 c-Met (Met1160)。此外,芳香杂环的侧链对分子与靶点的亲和力有显着影响;(ii) II区由吡啶环或苯环组成,其可以是未取代的或单取代的;(iii) 在区域 III 中,引入包含一个或多个氢键供体或受体的柔性链或刚性环结构(5 原子连接基团)提高了分子的效率。该区域与 VEGFR(Lys868、Asp1046 等)的氨基酸残基之间形成氢键。) 和 c-Met (Asp1220、Lys1110、Leu1245 等);(iv) IV 区由一个六元芳香杂环组成,它可以是未取代的、单取代的或双取代的[93 ]。在这里,我们总结了双重 VEGFR/c-Met 抑制剂的主要成就,它们的化学结构、效力和发展如图 1 和图 2 所示。 6和7。
表 3 临床批准的双重 VEGFR-c-Met 抑制剂总结
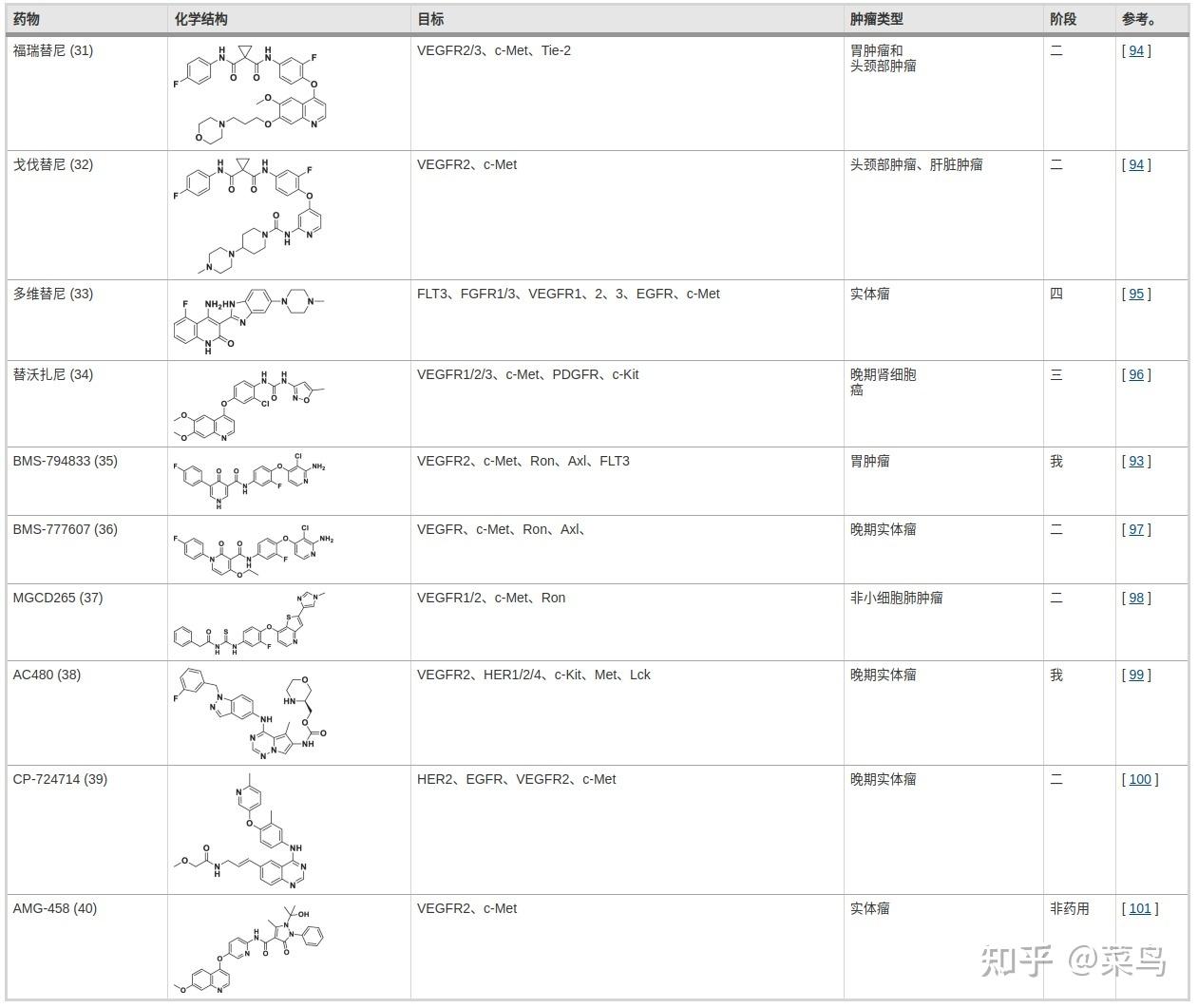
图五 VEGFR2/c-Met双重抑制剂的结构式
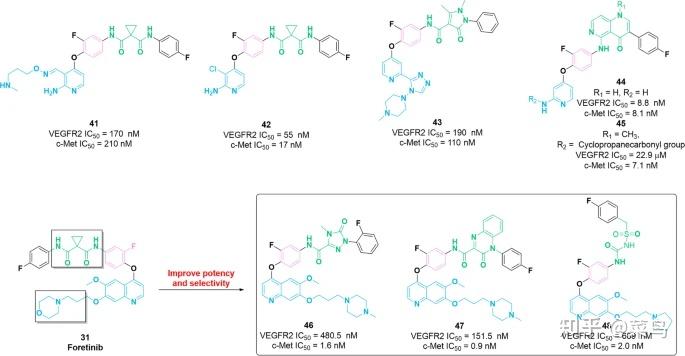
图六 双重 VEGFR-c-Met 抑制剂的化学结构和性质41 – 48
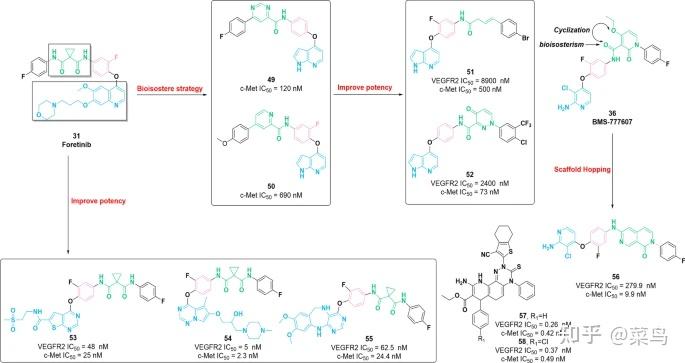
图七 VEGFR-c-Met 双重抑制剂的化学结构49-58及其对 VEGFR2 和 c-Met 的抑制活性
吡啶/嘧啶支架已广泛应用于包括 VEGFR 在内的 RTK 抑制剂。特别是,吡啶基序延伸到靶蛋白的 ATP 结合口袋中,并与铰链区中的相邻残基相互作用 [ 102 ]。2016 年,设计合成了一系列氨基嘧啶衍生物,以评估其对 VEGFR2 和 c-Met 的抑制活性[ 103 ]。在这些化合物中,分子41对 VEGFR2 和 c-Met 具有相当大的效力,IC 50值分别为 170 nM 和 210 nM。次年,赵等人。根据分子41的结构鉴定出化合物42,它是一种有效的 VEGFR2/c-Met 双重抑制剂 [ 104 ]。此外,42在体外对 VEGFR2 (IC 50 = 55 nM) 和 c-Met (IC 50 = 17 nM)具有细胞活性和强效。这些化合物的初步 SAR 表明,氯原子的引入可以正向调节对 VEGFR2 和 c-Met 的激酶抑制活性。同年,顾等人。设计并合成了一系列新型 VEGFR2/c-Met 双重抑制剂。其中,化合物43对靶标 [IC 50 = 160 nM (VEGFR2) 和 110 nM (c-Met)],与阳性对照6 [ 105 ] 相比,对人血管内皮细胞 HUVEC 和 BaF3-TPR-Met 细胞的抗增殖作用较差。对接研究进一步证实,分子43占据了 VEGFR2 和 c-Met 的 ATP 结合口袋,其吡啶和三唑部分与 VEGFR2 (Cys919) 和 c-Met (Tyr1159 和 Met1160) 的氨基酸残基形成氢键。此外,化合物43的 5 原子连接基团(吡唑啉酮部分)与 VEGFR2(Val898 和 Lys868)和 c-Met(Asp1222)的氨基酸残基产生至少一个氢键。类似地,使用脚手架跳跃策略,化合物44含有 1,6-萘啶支架被鉴定为有效的 VEGFR2 和 c-Met 双重抑制剂,IC 50值分别为 68 nM 和 9.8 nM [ 106 ]。此外,44具有不利的药代动力学特征(F % = 12,CL = 5.0 L/h/kg)。基于分子44的结构进行了进一步优化,因此,化合物45被鉴定为 c-Met 的有效抑制剂 (IC 50 = 7.1 nM),对 VEGFR2 具有选择性。PK 研究显示中等清除率 (CL = 0.02 L/h/kg) 和合适的口服生物利用度 (F = 57%) 为44. SAR 研究表明,2-氨基嘧啶骨架上的取代比 VEGFR2 对 c-Met 功效的抵抗力更强。
喹啉核心被广泛用于设计几种具有不同生物学特性的活性分子。2016 年,基于化合物31的 SAR 研究, Liu 等人确定分子46是 c-Met 的有效抑制剂(IC 50 = 1.57 nM) 。[ 107 ]。46对c-Met的选择性比VEGFR2高306倍,比PDGFRα和Ron高100倍。此外,46对不同类型的肿瘤细胞(HT-29、H460、A549 和 MKN-45)具有显着的抑制效力,IC 50值分别为 80 nM、140 nM、110 nM 和 30 nM。进一步的 SAR 研究表明,用 5-oxo-4,5-dihydro-1 H -1,2,4-triazole-取代31的 5 原子连接基团(环丙烷-1,1-二甲酰胺部分)3-甲酰胺片段和1,2,4-三唑骨架4位上的结构修饰(甲基、乙基或环丙基)是可以接受的。有趣的是,在末端苯环的 2 位引入氟原子将有利于有效的细胞毒性。类似地,3-氧代-3,4-二氢喹喔啉-2-甲酰胺部分表现出与环丙烷-1,1-二甲酰胺部分相似的性质,特别是含有氢键供体和受体。这些研究加速了化合物的鉴定47,其在 5 原子连接基团处含有 3-氧代-3,4-二氢喹喔啉-2-甲酰胺部分 [ 108 ]。 与分子31 (IC 50 = 1.41 nM ) 相比, 47对 c-Met 的抑制活性 (IC 50 = 0.9 nM) 更强。此外,47对 c-Kit 表现出高抑制效力 (IC 50 = 2.45 nM),对 Ron、VEGFR2 和 FLT3 具有相当大的功效,IC 50值分别为 82.56 nM、151.47 nM 和 268.81 nM。进一步的体外研究表明,47显示出显着的细胞毒性(IC 50纳摩尔浓度范围内的值)针对不同类型的肿瘤细胞。遗憾的是,缺乏体内研究。2019年,通过化合物31的结构优化,发现了含有4-苯氧基喹啉骨架和磺酰脲部分的c-Met抑制剂48 [ 109 ]。它对 c-Met 具有出色的抑制作用,IC 50值为 1.98 nM。此外,48对 c-Met 的选择性比 VEGFR 高 347 倍以上。48对不同类型的肿瘤细胞系具有很强的抗增殖活性,在体外具有纳摩尔效力。SAR 研究表明,引入磺酰脲片段作为 5 原子连接基团可以保持显着的效力。近年来,吡咯并吡啶衍生物作为生物活性分子,在药物化学中占有独特的地位[ 110 ]。朱等人。使用生物等排策略鉴定了化合物49和50作为 c-Met 的强效抑制剂(IC 50 = 120 nM 和 670 nM),其对不同类型的肿瘤细胞具有优异的细胞毒性活性 [ 111 , 112 ]。基于49和50的结构,化合物51含有一个 N-酰基腙基团,被 Wang 等人鉴定为潜在的 c-Met 抑制剂 (IC 50 = 0.5 μM) 。[ 113 ] 。此外,与其他激酶(FLT3、VEGFR2 和 EGFR)相比, 51对 c-Met 显示出相当大的选择性,并通过在 G2/M 期阻止细胞周期诱导细胞凋亡而对不同类型的肿瘤细胞发挥显着的抑制活性。类似地,同一团队通过将 4-氧代-哒嗪酮片段引入分子49的 5 原子连接基团中鉴定了化合物52。它对 c-Met 具有显着的抑制活性,IC 50值为 73 nM。的选择性c-Met 的52比 VEGFR2 和 c-Kit 高约 15 倍,具体而言,比 FLT3 高 7 倍。体外试验表明,分子52对不同类型的肿瘤具有良好的抑制活性 [ 114 ]。
同样,基于分子31的结构,制备了一系列噻吩并[2,3- d ]嘧啶衍生物作为 VEGFR2 和 c-Met 的有效双重抑制剂 [ 115 ]。在这些化合物中,分子53对 VEGFR2 和 c-Met 具有显着的抑制活性,IC 50值分别为 48 nM 和 25 nM。对接研究表明分子53的噻吩并[2,3- d ]嘧啶支架与 VEGFR2 (Cys919) 和 c-Met (Met1160) 的氨基酸残基产生氢键。此外,4-氟-苯基-环丙烷-1,1-二甲酰胺片段与 VEGFR (Asp1046 和 Lys868) 和 c-Met (Phe1223) 的氨基酸残基之间形成氢键。2018 年,含有吡咯并[1,2- f ][1,2,4]三嗪核心的化合物54被确定为 VEGFR2 (IC 50 = 5.0 nM) 和 c-Met (IC 50 = 2.3 nM)的显着双重抑制剂[ 116 ]。54对 BaF3-TPR-Met、HUVEC 和不同类型的肿瘤细胞显示出显着的抑制活性。此外,化合物54具有良好的理化性质和优异的药代动力学特征(F % = 98.1)。进一步的对接研究表明,分子54可以完全占据 c-Met 和 VEGFR2 的 ATP 结合口袋,从而产生重要的配体相互作用。同年,黄等人。将化合物55鉴定为 c-Met 抑制剂,它带有 6,11-dihydro-5 H - benzo[e]pyrimido[5,4 - b ] [ 1 , 4 ] 二氮杂类支架 [ 117 ]。酶抑制试验表明, 55对 c-Met 和 VEGFR2 具有显着的抑制效力,IC 50值分别为 24.4 nM 和 62.5 nM。化合物55对 VEGFR2 和 c-Met 的选择性高于其他激酶。此外,分子55具有良好的体外效力和中等的口服生物利用度 ( F % = 39)。进一步的体内证据表明,化合物55具有相当大的治疗效果(TGI = 64.5%)。
2019年,卓等人。通过基于知识和结构的方法,设计并合成了一系列基于 2,7-萘啶酮的 c-Met 抑制剂 [ 118 ]。体外试验表明,分子56作为最有希望的抑制剂,对 c-Met 具有强效(IC 50 = 9.9 nM),对 c-Met 的选择性比 VEGFR2 高 28 倍。此外,化合物56在小鼠异种移植肿瘤模型中具有良好的药代动力学特征 ( F % = 54) 和出色的体内功效 (TGI = 95%)。基于这些研究,2,7-萘啶酮可能是未来药物开发的有希望的骨架。2020 年,化合物57和58含有四氢苯并[ b ]噻吩的支架被鉴定为多靶点RTK抑制剂[ 119 ]。具体而言,57和58对包括 c-Met 和 VGEFR2 在内的多激酶具有显着的效力,其 IC 50值在低纳摩尔至皮摩尔浓度范围内。此外,它们还在体外对 A549、H460、HT-29、MKN-45、U87MG 和 SMMC-7721 等六种典型肿瘤细胞发挥抗增殖活性。然而,仍然缺乏体内研究。
双 VEGFR2-BRAF 抑制剂
正如我们之前提到的,涉及 VEGFR 抑制剂和 BRAF 抑制剂的联合治疗已被确定为一种有效的治疗策略 [ 120 ]。值得注意的是,RAF-265 是一种 VEGFR2-BRAF 双重抑制剂,已在 I/II 期临床试验(NCT00304525)中证明了其有效性和安全性。目前,已鉴定出几种双重 VEGFR2-BRAF 抑制剂可抑制肿瘤生长。它们的化学结构、体外和体内效力以及优化如图 8所示。
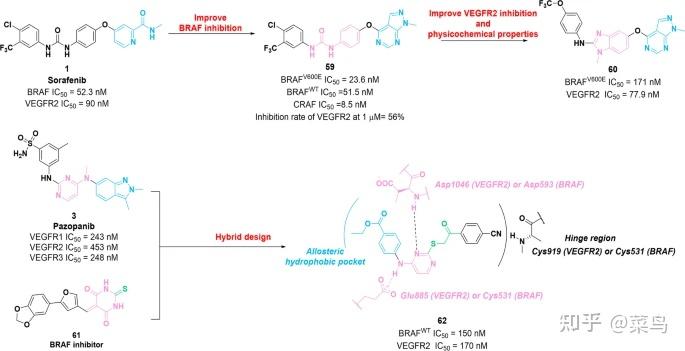
图八 VEGFR-BRAF 双重抑制剂的化学结构59、60和62及其对 VEGFR2 和 BRAF 的抑制活性
2017年,傅等人。通过使用基于结构的药物设计,将化合物59鉴定为令人鼓舞的 RAF 抑制剂 [ 121 ]。它对 c-RAF、野生型 BRAF 和 BRAF V600E具有显着的抑制效力,IC 50值分别为 8.5 nM、51.5 nM 和 23.6 nM。此外,59在体外对四种具有微摩尔效力的肿瘤细胞系具有很强的抗增殖活性,并且与其他激酶相比对 RAF 具有更高的选择性。特别是,59对 VEGFR2 激酶具有中等抑制活性。(1 μM 时 VEGFR2 的抑制率为 56%。)机理研究表明,59通过在 G0/G1 期阻滞细胞周期进程并显着抑制 RAS/RAF/MEK/MAPK 信号通路,对 A375 和 HT-29 细胞发挥抗增殖活性。次年,基于59的结构优化开发了化合物60,其对 VEGFR2 (IC 50 = 77.9 nM) 和 BRAF V600E (IC 50 = 171 nM) 的选择性优于野生型 BRAF 和其他蛋白激酶 [ 122 ]。对接研究和分子动力学模拟表明,60在 BRAF V600E的 ATP 结合位点采用与化合物1相似的结合模式和 VEGFR2。这些研究表明,60可以作为先导化合物用于鉴定有效的 BRAF V600E /VEGFR2 双重抑制剂。
2019 年,通过 VEGFR 抑制剂3和 BRAF 抑制剂61之间的结构杂交,化合物62被鉴定为有效的野生型 BRAF/VEGFR2 双重抑制剂[ 123 ]。62对野生型 BRAF 和 VEGFR2 具有有效的抑制活性,IC 50值分别为 150 nM 和 170 nM。此外,62对 MCF-7 和 T-47D 细胞具有抗增殖活性,IC 50值在微摩尔浓度范围内。对接研究表明,62可以与VEGFR2和BRAF的活性位点结合,从而完成关键的结合相互作用。
双 VEGFR2-HDAC 抑制剂
最近的研究表明,VEGFR 抑制剂和 HDAC 抑制剂的组合在体外和体内发挥了有希望的效力。在结构上,HDAC抑制剂通常由三部分组成:锌结合基团(ZBG)、适当的接头和封端基团(CAP组)。值得注意的是,SAR 研究表明,HDAC 抑制剂 CAP 组的修饰是可以耐受的。因此,CAP 组可以与 VEGFR2 抑制剂杂交以鉴定双靶点抑制剂 [ 124 ]。2016 年,在母体化合物4和63 (SAHA)的基础上,获得了一系列含有N-苯基喹唑啉-4-胺和异羟肟酸部分的 VEGFR2/HDAC 双抑制剂[ 125]。其中,分子64(图 9)对VEGFR2(IC 50 = 74 nM)和HDAC(IC 50 = 2.2 nM)具有显着的效力,对人乳腺肿瘤细胞MCF-7具有良好的抑制活性,IC 50值为850纳米。不幸的是,缺乏化合物64对 HDAC 家族成员的选择性特征。最近,通过结合 VEGFR 抑制剂1和 HDAC 抑制剂63的药效团,合成了一系列苯脲异羟肟酸,以评估它们对 VEGFR2 和 HDAC 的抑制活性 [ 126 ]。在这些化合物中,分子65(图 1)。 9 ) 有效抑制 HDAC6 (IC 50 = 166 nM),并且对 HDAC6 的选择性高于对其他 HDAC1、HDAC2 和 HDAC8 的选择性。此外,65对 VEGFR2 的效力较弱,IC 50值为 13.2 μM。1与 VEGFR2 复合物的共晶结构(PDB: 3EWH) 表明关键的N-甲基-2-吡啶甲酰胺基团对 VEGFR2 的效力具有重要意义,它通过与 VEGFR2 产生两个氢键插入铰链区Cys919 [ 127 ]。因此,对分子1的这个区域进行不合理的修饰会导致抑制活性的丧失。
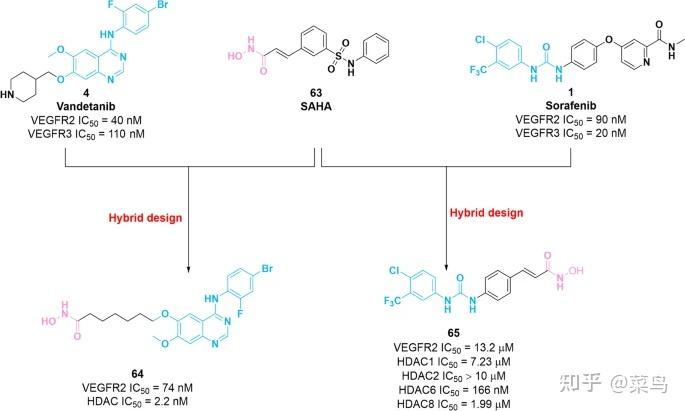
图九 VEGFR-HDAC双重抑制剂64和65的化学结构及其对VEGFR2和HDAC的抑制活性
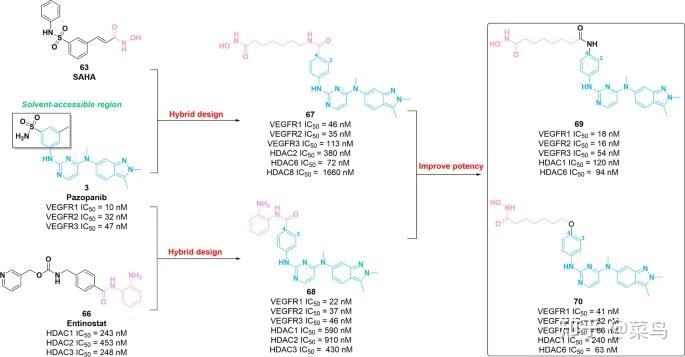
2018年,张等人。使用 VEGFR 抑制剂3的药效团作为 CAP 组和不同的接头组、异羟肟酸或邻氨基苯胺作为 ZBG,产生了两个系列的双重 VEGFR-HDAC 抑制剂 [128]。其中,化合物67和68(图 10)对VEGFR和HDAC发挥有效效力,IC50值在纳摩尔或低微摩尔浓度范围内。具体而言,与分子63相比,化合物67发挥了相当大的 HDAC2/6 抑制效力和优越的 HDAC8 抑制作用。分子68与分子66(Entinostat)相比,对 HDAC1/2/3 也具有相当的功效。此外,与化合物3(VEGFR2 IC50 = 32 nM) 相比,67和68对 VEGFR2 显示出相似的抑制活性,IC50值分别为 35 nM 和 37 nM。其他激酶(VEGFR1、VEGFR3、PDGFRβ、FGFR、C-Fms 和 c-Kit)是受3抑制的肿瘤相关靶标,可被分子67和68有效抑制。SAR 为67和68可简要总结如下:(i)分子3的溶剂暴露苯基部分的结构修饰因其激酶抑制特性而具有良好的耐受性;(ii) 溶剂暴露的苯基的 4 位取代有利于对抗 HDAC 和 VEGFR2 的效力。此外,化合物68在 HT-29 异种移植模型中表现出理想的药代动力学特征 (F% = 72) 和适度的体内抗肿瘤作用 (TGI = 40%)。2022年,在67结构优化的基础上,开发出化合物69和70(图 11 ),与分子67[129]相比,其对 HDAC 的效力和对 VEGFR 的抑制活性更强。此外,分子69和70对不同类型的肿瘤细胞显示出良好的抗增殖活性,IC50值在微摩尔浓度范围内。
图十 VEGFR-HDAC双重抑制剂的化学结构67-70及其对VEGFR2和HDAC的抑制活性
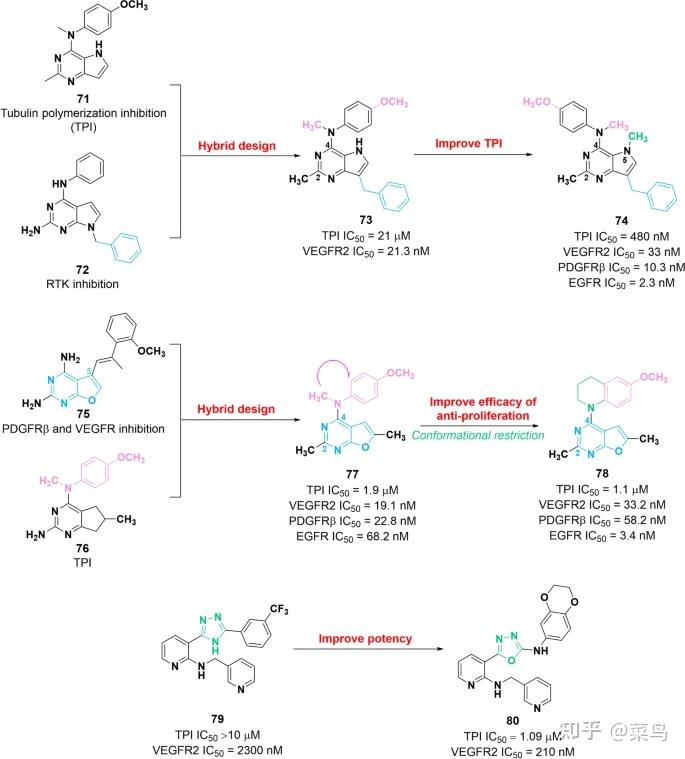
图十一 VEGFR-微管蛋白双重抑制剂 73、74、77、78、79 和 80 的化学结构及其对VEGFR2和微管蛋白聚合( TPI) 的抑制活性
双 VEGFR2-微管蛋白抑制剂
微管蛋白抑制剂和 VEGFR2 抑制剂联合治疗的效果已被多项研究证实 [ 130 , 131 ]。目前,已鉴定出几种双重 VEGFR2-微管蛋白抑制剂发挥有效的抗肿瘤活性。它们的化学结构、体外效力和优化示于图 11。
2014年,通过将RTK抑制剂72的7-苄基部分引入微管蛋白抑制剂71的核心,制备了一系列VEGFR2-微管蛋白抑制剂[ 132 ]。其中,分子73对 VEGFR2 和微管蛋白聚合具有显着的抑制作用,IC 50值分别为 21.3 nM 和 21 μM。初步 SAR 研究表明 (i) 7-苄基片段在维持针对 VEGFR2 的效力中起关键作用;(ii) N-CH 3和O-CH 3对VEGFR2 和微管的抑制活性是必不可少的。体外试验表明,73具有强大的抗血管生成和抗增殖活性。具体来说,73对过表达 βIII-微管蛋白的 HeLa 细胞 (IC 50 = 280 nM) 和过表达 P-gp 的 ADR-RES 细胞 (IC 50 = 700 nM) 具有显着的抑制活性,因此理论上可以逆转 βIII-微管蛋白和 P- gp过表达诱导的耐药性。此外,73在体内肿瘤模型中显示出有效的抗肿瘤和抗转移作用。2017年,基于73的结构开发了化合物74,对VEGFR2(IC 50 = 33 nM)、微管蛋白聚合(IC 50 = 480 nM)、EGFR(IC 50 = 2.3 nM) 和 PDGFRβ (IC 50 = 10.3 nM) 在体外 [ 133 ] 。此外,与73相比, 74对 βIII-微管蛋白过表达 (IC 50 = 250 nM) 和 P-gp 过表达 (IC 50 = 70 nM) 的肿瘤细胞具有更好的细胞毒性活性。进一步的 SAR 研究表明,N4-CH 3和 N5-CH 3基团在抑制微管蛋白聚合的效力中起关键作用。在结构上,N5-CH 3基团被认为有利于构象刚性的形成,从而提高疗效。此外,2-CH 3基团被2-氨基部分取代,导致对微管蛋白聚合的抑制活性降低。
在另一份报告中,RTK 抑制剂75和微管蛋白抑制剂76的药效团在单个分子中的杂交促进了化合物77 [ 134 ] 的发现。该分子对 EGFR、VEGFR2、PDGFRβ 和微管蛋白聚合具有良好的效力,IC 50值分别为 68.2 nM、19.1 nM、22.8 nM 和 1900 nM。此外,体外和体内证据证明,与多西紫杉醇相比,化合物77在增殖抑制和抑制肿瘤血管生成方面的效力更高。基于化合物77的结构和进一步的配体设计,化合物78进一步发现对于微管蛋白聚合 (IC 50 = 1100 nM) 和 EGFR (IC 50 = 3.4 nM) [ 135 ] ,化合物77更有效。然而,78对 VEGFR2 (IC 50 = 33.2 nM) 和 PDGFRβ (IC 50 = 58.2 nM) 的效力较差。SAR研究表明,四氢喹啉环片段的引入有利于改善EGFR抑制。此外,78显着抑制耐药性 HeLa 和 SK-OV-3 细胞的生长,IC 50值分别为 9.1 nM 和 19.4 nM。
含有 1,3,4-恶二唑片段的化合物80是基于弱 VEGFR2 抑制剂79的结构优化开发的,与微管蛋白聚合 (TPI IC 50 > 10 μM) 和 VEGFR2 (IC 50 = 2300 nM) 相比,它具有有希望的效力与分子80相同(TPI IC 50 = 1090 nM,VEGFR2 IC 50 = 210 nM)[ 136 ]。此外,80可以阻断 G2/M 期的细胞周期进程。急性和重复剂量口服毒性研究表明,80具有良好的安全性。
VEGFR2-ERα双重抑制剂
SERM 和 VEGFR 抑制剂的联合治疗已被确定为延缓 SERM 耐药性肿瘤生长的有效治疗策略 [ 137 ]。获得了许多具有显着抗乳腺肿瘤活性的双重VEGFR2 -ERα 抑制剂。它们的化学结构、体外效力和优化如图 12所示。
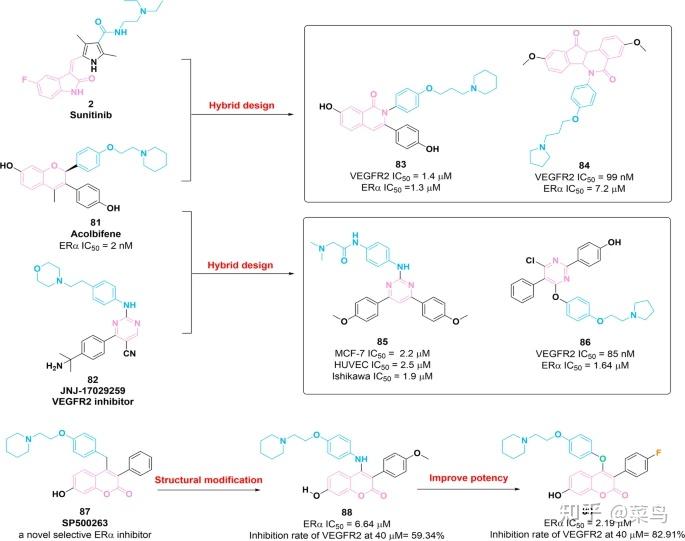
图十二 VEGFR-ERα双重抑制剂83-86、88和89的化学结构及其对VEGFR2和ERα的抑制活性
2014 年,基于 VEGFR 抑制剂2和 SERM 81 (acolbifene) 的结构,进一步发现化合物83对 ERα (IC 50 = 1.3 μM) 和 VEGFR2 (IC 50 = 1.4 μM) 具有相当强的效力 [ 138 ]。生物学研究表明,83对 MCF-7 细胞具有抗增殖活性,IC 50值为 2.73 μM,并具有潜在的体内抗血管生成功效。2016年,基于2和81的结构制备了一系列含有芳基-茚并异喹诺酮核心的VEGFR2/ERα抑制剂[ 139 ]。模拟84获得,显示出对 VEGFR2 和 ERα 的显着效力,IC 50值分别为 99 nM 和 7.2 μM。此外,84对 MCF-7、MDA-MB-231、Ishikawa 和 HUVEC 细胞系具有良好的细胞毒性活性,IC 50值分别为 1.2 μM、0.5 μM、8.2 μM 和 800 nM。进一步的体外研究表明,84通过负调控 VEGFR2 和 RAF-1/MAPK/ERK 通路的信号转导抑制 MDA-MB-231 细胞的生长。
2017年,通过81和82的生物活性药效团的杂交,含有4,6-二芳基-2-嘧啶胺支架的化合物85被报道为潜在的乳腺肿瘤治疗药物。它对 MCF-7、HUVEC 和 Ishikawa 细胞具有良好的抑制活性 [ 140 ]。鸡绒毛尿囊膜 (CAM) 测定表明85在体内具有显着的抗血管生成活性。基于分子85的结构进行了进一步优化,因此,化合物86被确定为 VEGFR2 (IC 50 = 85 nM) 和 ERα (IC 50 = 1.64 μM) 的有效抑制剂 [ 141]。此外,86通过下调 MCF-7 细胞中孕酮受体 (PgR) mRNA 的表达具有显着的抗雌激素特性,并在体外和体内发挥显着的抗血管生成功效。
化合物87 (SP500263) 是一种基于香豆素的 SERM,对 ERα 具有高亲和力,并显着抑制雌激素依赖性 MCF-7 细胞的生长 [ 142 ]。2017年,基于87的结构优化开发了化合物88,对ERα具有潜在效力(IC 50 = 6.64 μM),对VEGFR2的抑制活性较弱[ 143 ]。 为了提高效力,已鉴定出对 ERα 具有有利效力的分子89 (IC 50 = 2.19 μM) [ 143]。SAR研究表明,在香豆素核心的4位引入生物等排O原子对于增强ERα抑制是必不可少的。与分子88相比,89对MCF-7和Ishikawa细胞具有更优异的抑制活性。在 MCF-7 细胞中,89通过负调控 VEGFR2 的表达和 RAF-1/MAPK/ERK 通路的信号转导,诱导细胞凋亡和延长 G0/G1 期,抑制增殖和迁移。总的来说,VEGFR2/ERα抑制剂的结构特征在于存在芳香族支架和末端带有叔胺取代基的柔性侧链。上述两种药效团的引入有利于对ERα和VEGFR的抑制活性。
双 VEGFR2–PIM1 抑制剂
PIM-1 激酶的表达已被认为是对 VEGFR 抑制剂的一种新的耐药机制 [ 144 ]。因此,涉及 PIM1 激酶和 VEGFR 抑制剂的联合治疗已被确定为使肿瘤细胞敏感的有效治疗策略。2019年,通过VEGFR抑制剂(化合物5和90)与PIM1抑制剂(图 13)的分子杂交制备了一系列含有噻吩并[2,3- b ]吡啶核心的PIM1/VEGFR2双重抑制剂(图13)[ 145 ]。在这些化合物中,发现91种对 PIM1 和 VEGFR2 具有最有效的抑制活性,IC 50值分别为 5873 nM 和 7948 nM。体外试验表明,91对不同类型的肿瘤细胞(HepG-2、Caco-2、MCF-7 和 PC-3)具有抑制效力,IC 50值在纳摩尔浓度范围内。此外,91可以正向调节caspase 3/7的表达并诱导肿瘤细胞凋亡。实时 PCR 分析表明,与多柔比星相比, 91在调节 VEGF、p53 和细胞周期蛋白 D 的表达方面表现出更好的治疗潜力(图 13)。
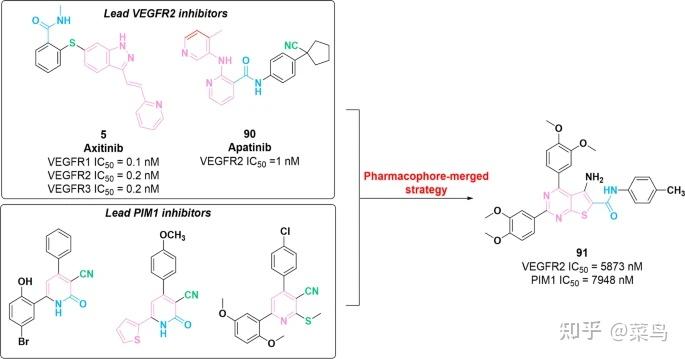
图十三 VEGFR-PIM1双重抑制剂91的化学结构及其对VEGFR2和PIM1的抑制活性
VEGFR2 和其他抗肿瘤靶点的双重抑制剂
目前,通过偶然发现或使用典型的设计策略鉴定了几种 VEGFR2 和其他抗肿瘤靶点的双重抑制剂,它们对相应的单靶点分子发挥了优越的效力。这些双重抑制剂经常被用作研究 VEGFR2 和其他抗肿瘤靶点协同相互作用的工具化合物。此外,它们还可以作为潜在的新线索来发现新的双靶点抗肿瘤药物。
在正常生理条件下,转染过程中重排(RET)在肾脏和神经系统的发育中起重要作用。在病理条件下,RET重排导致嵌合基因的产生。从机制上讲,这些基因是由 RET 酪氨酸激酶结构域与其他基因的 N 末端区域融合而成的。在结构上,VEGFR2 和 RET 在其 ATP 结合位点方面具有高度相似性。因此,几种靶向VEGFR 、 RET和其他激酶的多激酶抑制剂被广泛应用于临床,包括1、4、6和7。为了提高选择性和减少副作用,Brendan 等人。通过基于片段的化学筛选发现了一种双泛 RET/VEGFR2 激酶抑制剂92 (Pz-1),它具有显着的抑制活性,对 RET、VEGFR2 和 RET V804M具有纳摩尔效力。值得注意的是,体内结果证实了92在植入 RET 或 RAS 转化的 NIH3T3 成纤维细胞的裸鼠中具有良好的安全性和显着的肿瘤生长抑制作用[ 146 ]。2020年,为进一步增强92的代谢稳定性,通过对92易脱甲基位点进行生物等排取代,鉴定出化合物93(NPA101.3)(图14 ))。酶抑制试验表明,93对 RET 和 VEGFR2 均具有显着的抑制效力,IC 50值分别为 1 nM 和 3 nM。此外,体外研究表明,93可以抑制 RET 癌蛋白和 VEGFR2 的磷酸化。它还显着抑制 RET 转化的 Ba/F3 细胞的增殖,IC 50值在低纳摩尔浓度范围内。体内证据证明,化合物93完全阻止了由 RET C634Y转化细胞诱导的肿瘤形成 [ 147 ]。
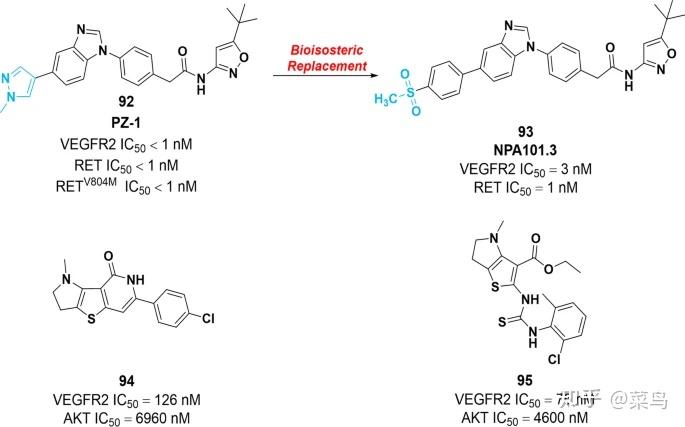
图十四 VEGFR-RET 抑制剂92、93和VEGFR-AKT 抑制剂94的化学结构及其对靶蛋白的抑制活性
VEGF与VEGFR结合可导致AKT活化,提高肿瘤细胞的增殖、迁移和侵袭能力。此外,一些研究表明,对 VEGFR 抑制剂的耐药性是导致 AKT 获得性突变的原因。因此,VEGFR2 和 AKT 的双重抑制可能会在不同的焦点处引发细胞凋亡。2022年,Abdelnaby等人制备了一系列含有噻吩并吡咯或吡咯噻吩并嘧啶支架的VEGFR-AKT双重抑制剂。其中,化合物94和95对AKT(IC 50 分别为6.96 μM和4.60 μM)和VEGFR2(IC 50 = 126 nM和75 nM)表现出更好的抑制活性(图14)。在 HepG2 细胞中,94和95可通过抑制 S 期细胞增殖和阻止细胞生长而加剧细胞凋亡,导致细胞凋亡 [ 148 ]。
结论和未来方向
目前,基于抑制VEGFR的抗血管生成疗法被认为是治疗实体瘤的有效临床策略。尽管VEGFR抑制剂在临床应用中显示出前瞻性疗效,但仍存在临床疗效中等、机制相关毒性和临床耐药发生等障碍和挑战。令人鼓舞的是,由于结构生物学和药物化学技术的进步,在确定新的联合治疗策略方面取得了巨大进展。多靶点,尤其是双靶点药物设计,是肿瘤治疗中最热门的领域之一。与联合化疗相比,多靶点药物具有协同抗肿瘤作用和改善药代动力学特性的优点。鉴于VEGFR在肿瘤血管生成发展中的关键作用,VEGFR的双靶点药物设计已成为药物研发领域的热门话题。多项研究证明,VEGFR 抑制剂和其他肿瘤相关靶点(包括 EGFR、FGFR、BRAF、c-Met、HDAC、微管蛋白、ERα 和 PIM1)抑制剂联合治疗肿瘤患者具有良好的疗效和安全性。
一般来说,混合设计策略将 VEGFR 抑制剂的活性基团与另一种肿瘤相关靶点抑制剂的药效团整合到一个分子中,以识别新的有效药物。在这篇综述中,我们总结了基于 VEGFR 的双靶点抑制剂,为未来设计涉及 VEGFR 的双靶点抑制剂提供了依据。临床实践和研究表明,VEGFR 抑制剂与其他肿瘤相关靶点的各种抑制剂具有协同作用 [ 149 ]。然而,双靶点药物设计方法尚未广泛应用于几个靶点,例如聚 ADP-核糖聚合酶 (PARP),它与 VEGFR 抑制剂具有协同作用 [ 150]。值得注意的是,多项临床研究证实了基于 VEGFR 的双靶点药物(如化合物23和26)治疗不同类型肿瘤的疗效和安全性。上述研究证实了基于 VEGFR 的双靶点药物设计策略的可行性。
然而,哪里有机会,哪里就有挑战。首先,根据报道的靶点与肿瘤之间的相关性确定合理的靶点组合是确定双靶点 VEGFR 抑制剂的主要挑战。如今,这通常是通过临床研究和基于表型的联合治疗筛查来实现的。此外,双靶点 VEGFR 抑制剂的临床成功取决于疗效、药代动力学特性和毒性的优化。为了满足这些需求,获得具有优异药代动力学特性的高效双靶先导化合物可以作为起点。更好的方法是最大化母体分子的药效团的重叠,以生成具有所需功能的更小分子,这些分子具有足够的化学空间进行结构优化。具体而言,在优化双靶点 VEGFR 抑制剂的药代动力学特性时,保持低亲脂性和避免过度的结构扩大是需要考虑的主要问题。活性母体分子的药效团具有高度的结构相似性。但是,药效团合并得到的双靶点分子不一定有效。其次,临床研究中的大多数强效VEGFR抑制剂都是多靶点的,例如化合物 药效团合并得到的双靶点分子不一定有效。其次,临床研究中的大多数强效VEGFR抑制剂都是多靶点的,例如化合物 药效团合并得到的双靶点分子不一定有效。其次,临床研究中的大多数强效VEGFR抑制剂都是多靶点的,例如化合物1 - 12。值得注意的是,这些药物由于选择性差、潜在毒性或代谢稳定性低等原因在一定程度上受到限制,严重影响了其临床应用。因此,迫切需要开发高选择性的 VEGFR 抑制剂。虽然高效、选择性的单靶点药物可以暂时解决这些问题,但这些药物受限于代偿性信号通路激活引起的耐药性。一种优越的方法是鉴定具有良好选择性和双重抑制效力的双靶点 VEGFR 抑制剂,同时抑制至少两个协同靶点。
令人欣慰的是,除了上述传统的药物发现策略外,许多新方法已被用于合理有效地设计双功能抑制剂的药物。特别是,基于计算的方法为开发新的双靶点 VEGFR 抑制剂提供了机会。这些策略通过预测 VEGFR 活性位点与其他肿瘤相关靶点之间的结构相似性或相关信号通路的可靠分析,促进识别双靶点 VEGFR 抑制剂的潜在合理靶点组合。此外,基于结构和配体的药物设计(SBDD 和 LBDD)已广泛应用于含有新型支架的双靶先导化合物的开发和双靶抑制剂的分子优化 [ 151]。值得注意的是,人工智能 (AI) 是药物发现的新兴趋势。随着人工智能技术的发展,高质量数据集、新假设和机器学习模型、新算法等多种方法被开发并应用于双靶点VEGFR抑制剂的鉴定[ 152 ]。最后,结构生物学领域遇到了无数的技术突破。因此,最近获得了许多配体-蛋白质复合物的高分辨率结构,并全面概述了配体-蛋白质相互作用的分子机制。这些发现通过基于结构的药物发现提供了对结构修饰的深入了解,并为双靶点抑制剂的鉴定提供了结构基础。
总的来说,我们强调了在开发双靶点 VEGFR 抑制剂以评估相关靶点的生理功能和病态影响方面取得的进展,并讨论了在发现和合理设计更有效的双靶点抑制剂方面的挑战和未来方向。 |
|



 发表于 2022-11-27 17:57:24
发表于 2022-11-27 17:57:24